We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
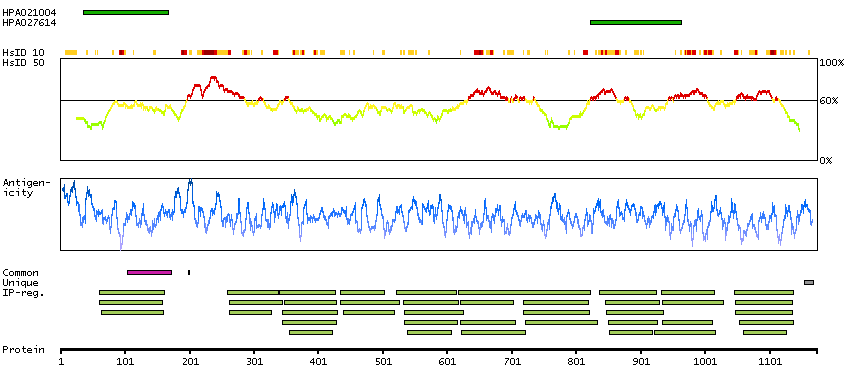
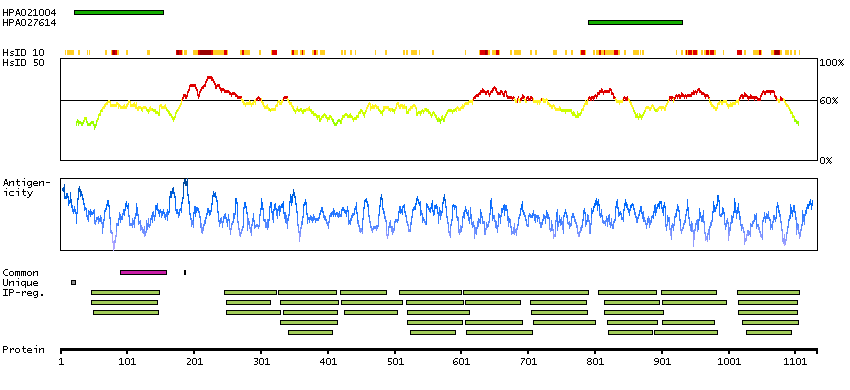
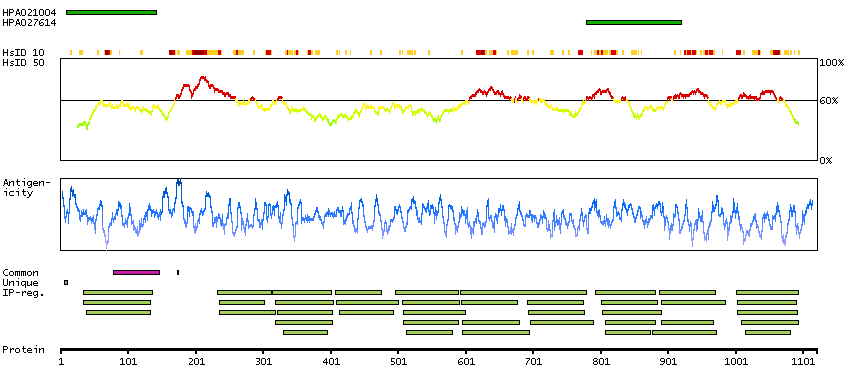
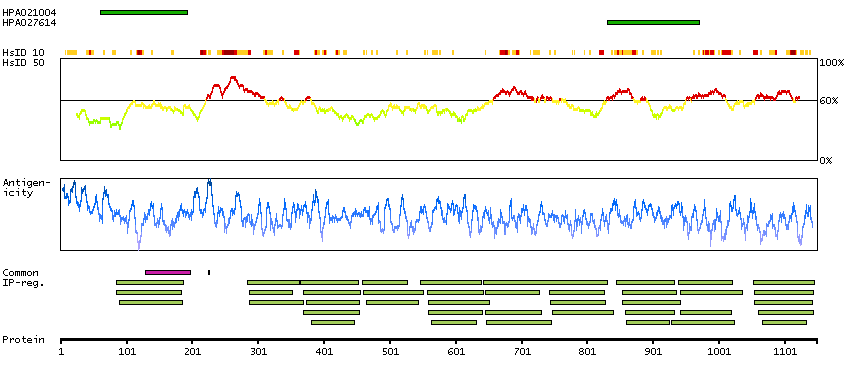
This gene encodes a member of the myosin-binding protein C family. Myosin-binding protein C family members are myosin-associated proteins found in the cross-bridge-bearing zone (C region) of A bands in striated muscle. The encoded protein is the slow skeletal muscle isoform of myosin-binding protein C and plays an important role in muscle contraction by recruiting muscle-type creatine kinase to myosin filaments. Mutations in this gene are associated with distal arthrogryposis type I. Alternatively spliced transcript variants encoding multiple isoforms have been observed for this gene. [provided by RefSeq, Dec 2011]