We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cytoplasmic expression in subsets of immune cells in several tissues, most abundant in bone marrow and spleen.
DATA RELIABILITY
Data reliability description
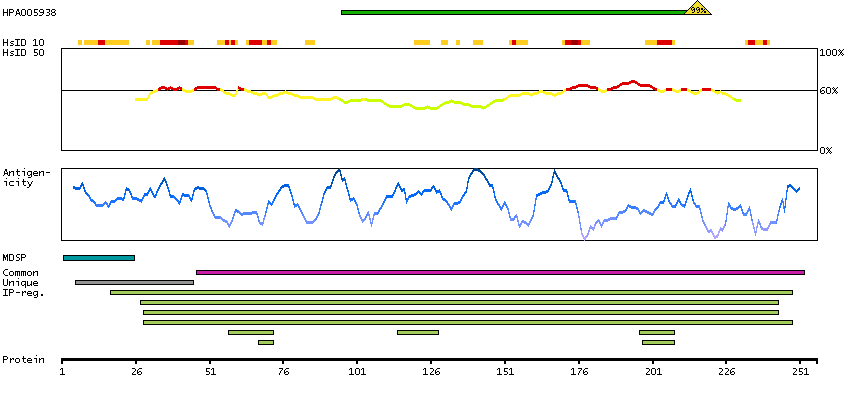
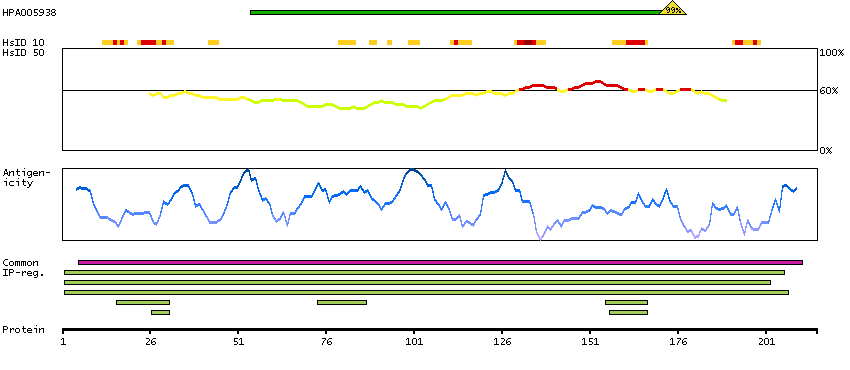
Antibody staining mainly consistent with RNA expression data. Presumed off target binding observed and disregarded. Antibody staining in cells/structures not annotated, view images.