We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes an enzyme which oxidizes carotenoids such as beta-carotene during the biosynthesis of vitamin A. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jan 2012]
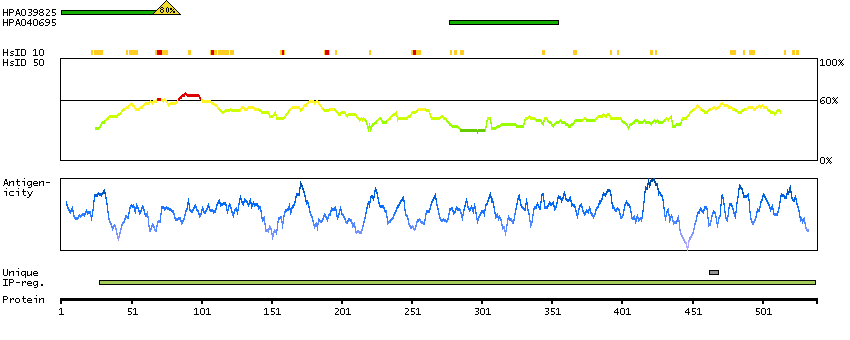
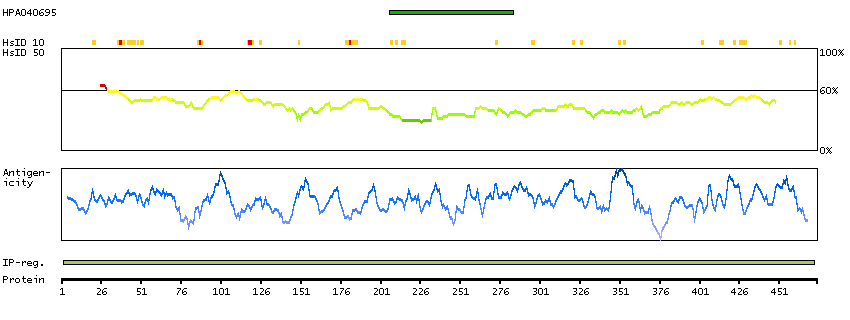
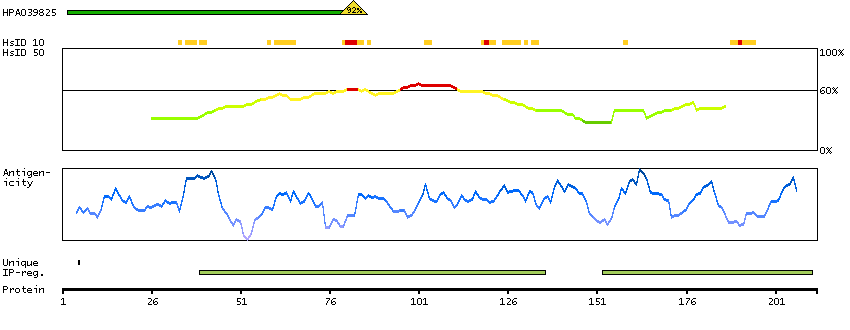
Enzymes ENZYME proteins Oxidoreductases SignalP predicted secreted proteins Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001523 [retinoid metabolic process] GO:0005622 [intracellular] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0007603 [phototransduction, visible light] GO:0016116 [carotenoid metabolic process] GO:0016119 [carotene metabolic process] GO:0016121 [carotene catabolic process] GO:0016122 [xanthophyll metabolic process] GO:0016702 [oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen] GO:0042573 [retinoic acid metabolic process] GO:0042574 [retinal metabolic process] GO:0046872 [metal ion binding] GO:0051881 [regulation of mitochondrial membrane potential] GO:0055114 [oxidation-reduction process] GO:2000377 [regulation of reactive oxygen species metabolic process]
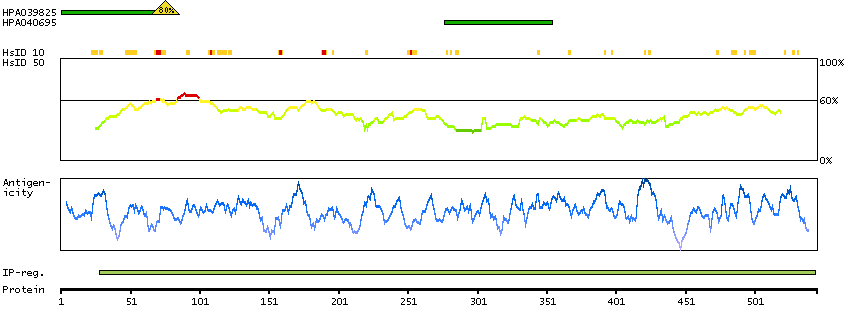
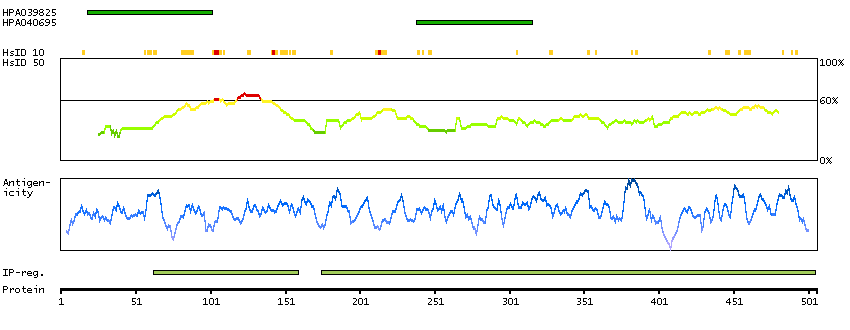
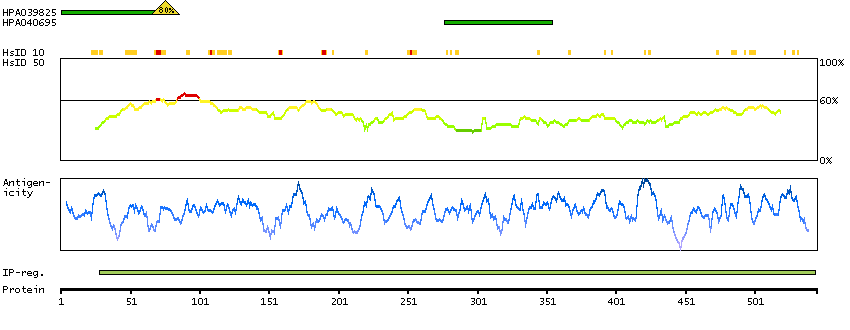
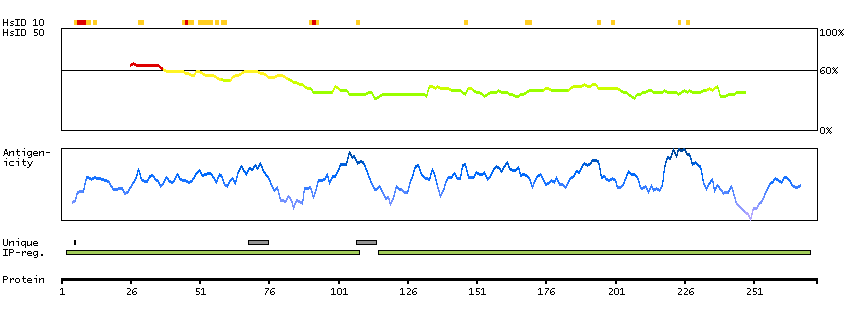
Enzymes ENZYME proteins Oxidoreductases SignalP predicted secreted proteins Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001523 [retinoid metabolic process] GO:0005622 [intracellular] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0007603 [phototransduction, visible light] GO:0016116 [carotenoid metabolic process] GO:0016119 [carotene metabolic process] GO:0016121 [carotene catabolic process] GO:0016702 [oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen] GO:0042573 [retinoic acid metabolic process] GO:0042574 [retinal metabolic process] GO:0046872 [metal ion binding] GO:0051881 [regulation of mitochondrial membrane potential] GO:0055114 [oxidation-reduction process] GO:2000377 [regulation of reactive oxygen species metabolic process]