We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
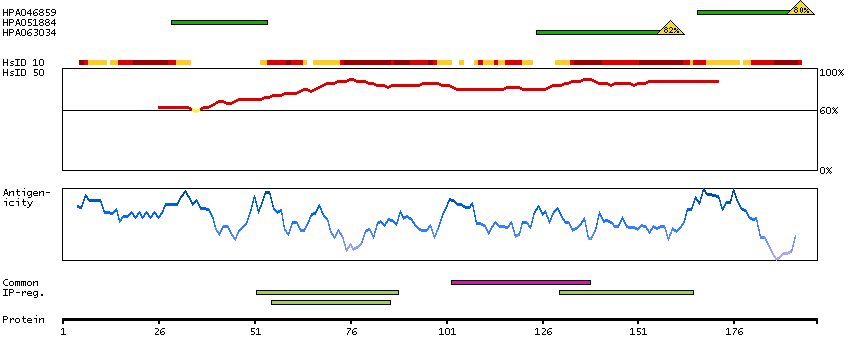
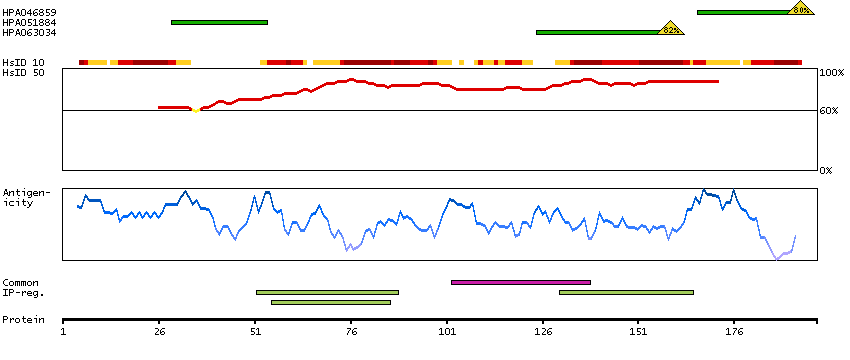
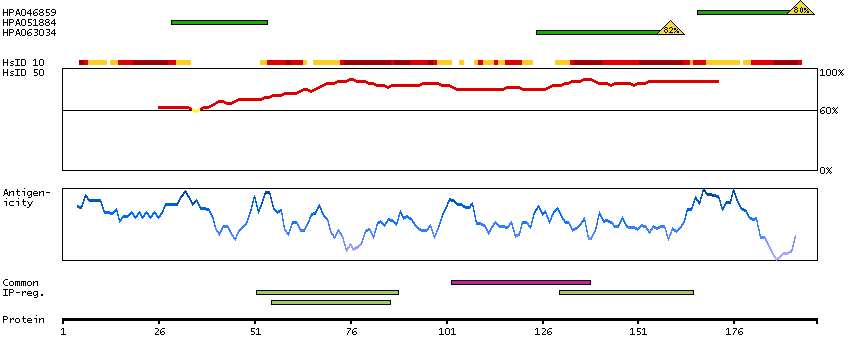
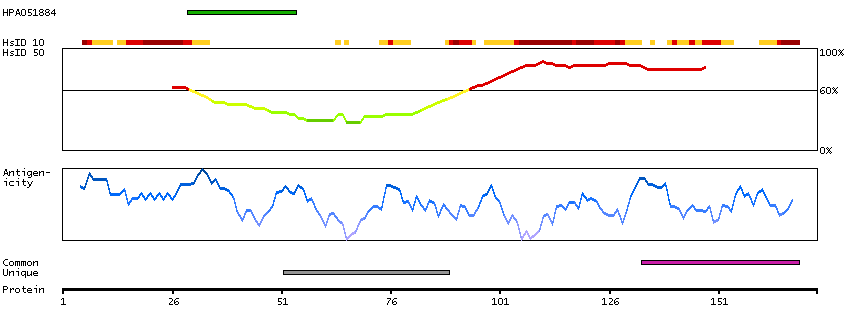
Myosin is a hexameric ATPase cellular motor protein. It is composed of two myosin heavy chains, two nonphosphorylatable myosin alkali light chains, and two phosphorylatable myosin regulatory light chains. This gene encodes a myosin alkali light chain that is found in embryonic muscle and adult atria. Two alternatively spliced transcript variants encoding the same protein have been found for this gene. [provided by RefSeq, Jul 2008]