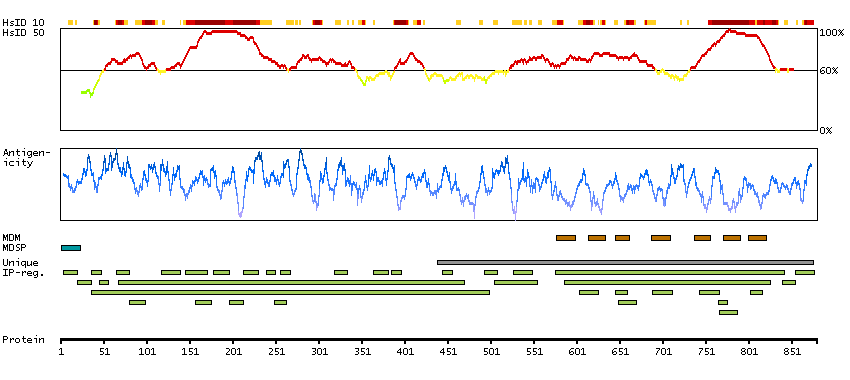
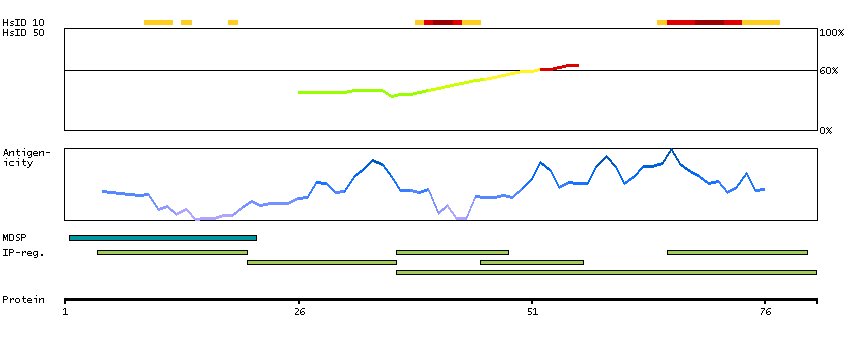
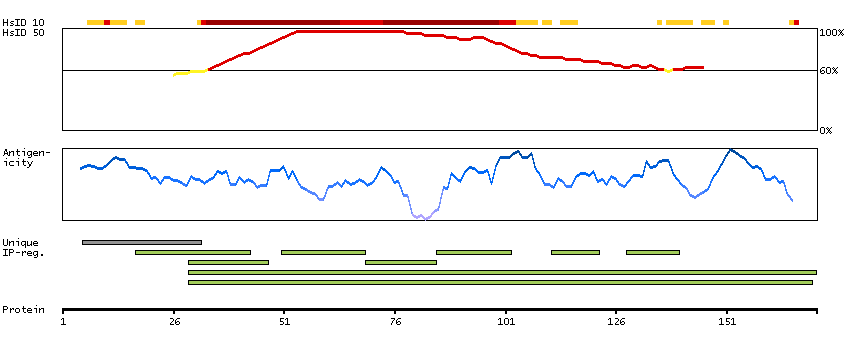
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (caudate, brain, hippocampus, cerebellum)
GENE INFORMATION
Gene name
GRM3 (HGNC Symbol)
Synonyms
GPRC1C, mGlu3, MGLUR3
Description
Glutamate receptor, metabotropic 3 (HGNC Symbol)
Entrez gene summary
L-glutamate is the major excitatory neurotransmitter in the central nervous system and activates both ionotropic and metabotropic glutamate receptors. Glutamatergic neurotransmission is involved in most aspects of normal brain function and can be perturbed in many neuropathologic conditions. The metabotropic glutamate receptors are a family of G protein-coupled receptors, that have been divided into 3 groups on the basis of sequence homology, putative signal transduction mechanisms, and pharmacologic properties. Group I includes GRM1 and GRM5 and these receptors have been shown to activate phospholipase C. Group II includes GRM2 and GRM3 while Group III includes GRM4, GRM6, GRM7 and GRM8. Group II and III receptors are linked to the inhibition of the cyclic AMP cascade but differ in their agonist selectivities. [provided by RefSeq, Jul 2008]