We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
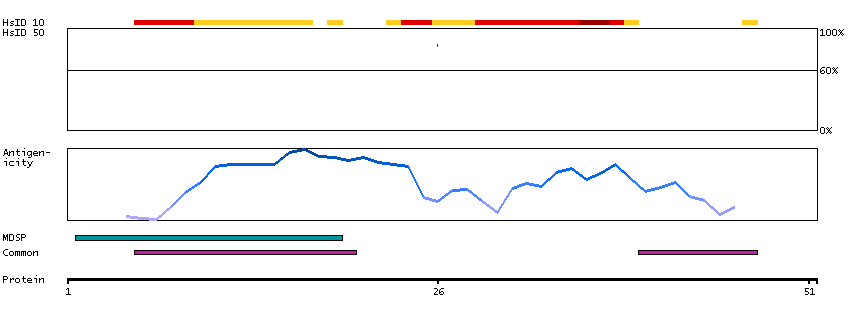
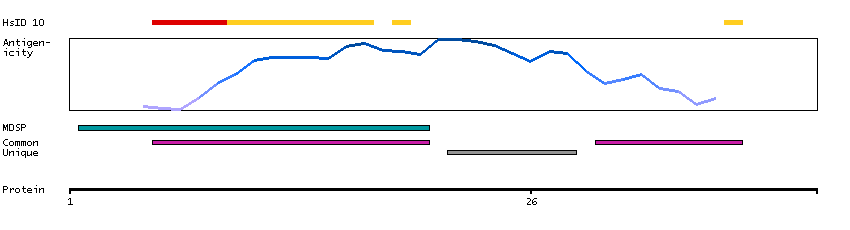
This gene encodes a member of the histatin family of small, histidine-rich, cationic proteins. They function as antimicrobial peptides and are important components of the innate immune system. Histatins are found in saliva and exhibit antibacterial, antifungal activities and function in wound healing. [provided by RefSeq, Sep 2014]