We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
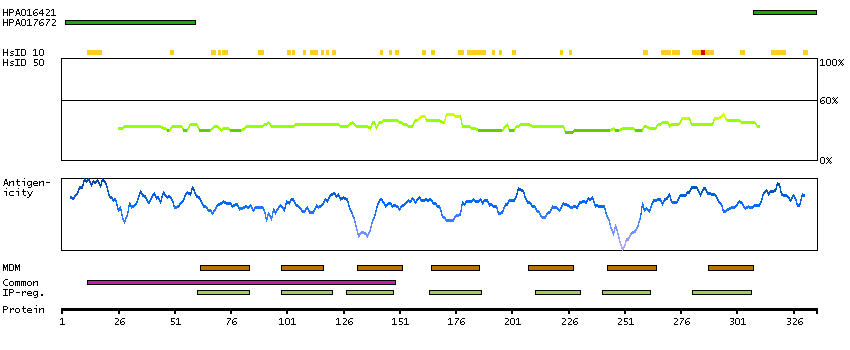
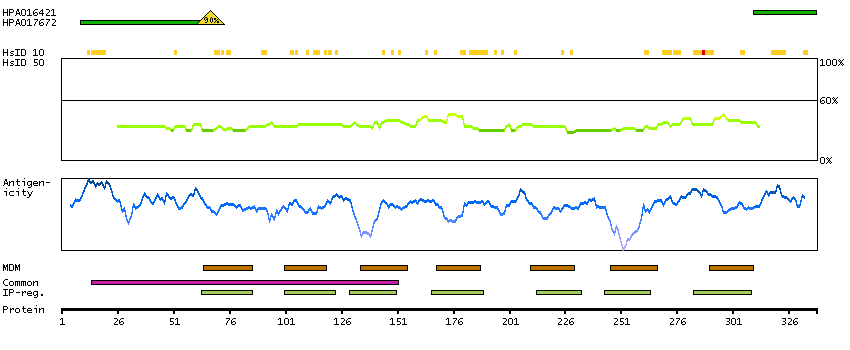
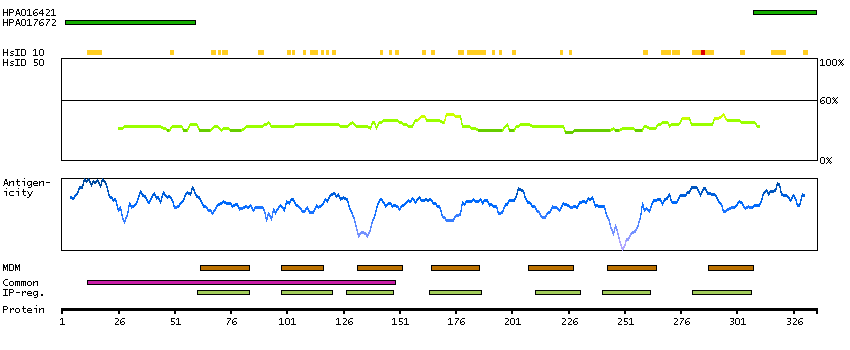
The protein encoded by this gene is a glycosylated membrane protein and a non-specific receptor for several chemokines. The encoded protein is the receptor for the human malarial parasites Plasmodium vivax and Plasmodium knowlesi. Polymorphisms in this gene are the basis of the Duffy blood group system. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]