We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
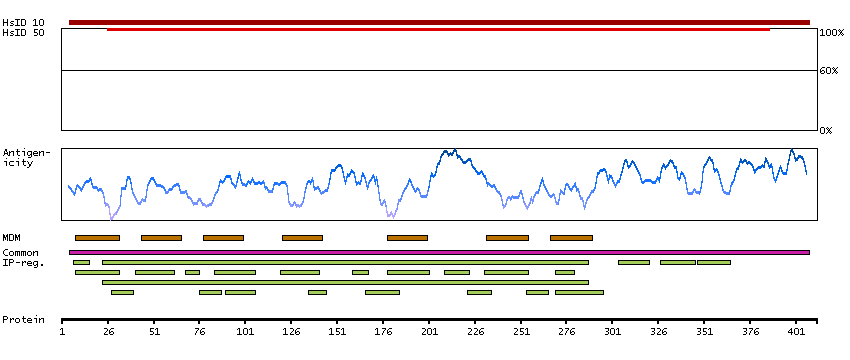
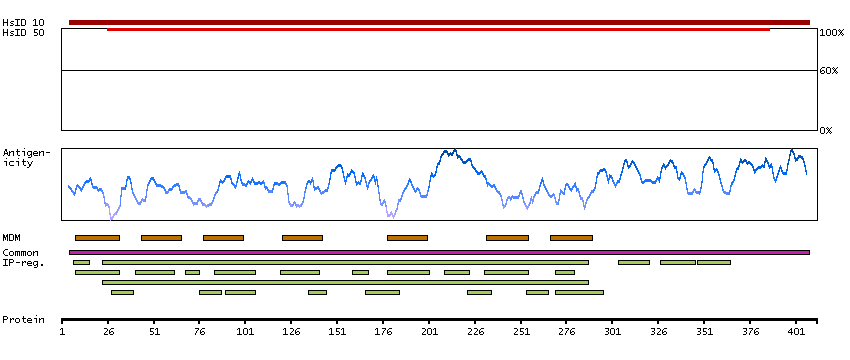
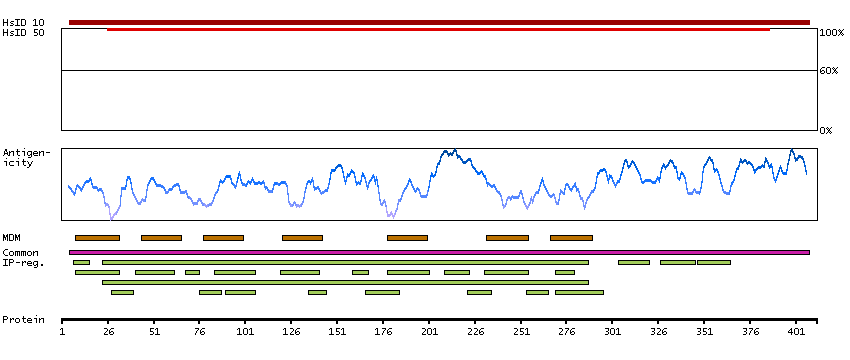
This gene encodes a member of the guanine nucleotide-binding protein (G protein)-coupled receptor (GPCR) superfamily, which is subdivided into classes and subtypes. The receptors are seven-pass transmembrane proteins that respond to extracellular cues and activate intracellular signal transduction pathways. This protein, an adenosine receptor of A2A subtype, uses adenosine as the preferred endogenous agonist and preferentially interacts with the G(s) and G(olf) family of G proteins to increase intracellular cAMP levels. It plays an important role in many biological functions, such as cardiac rhythm and circulation, cerebral and renal blood flow, immune function, pain regulation, and sleep. It has been implicated in pathophysiological conditions such as inflammatory diseases and neurodegenerative disorders. Alternative splicing results in multiple transcript variants. A read-through transcript composed of the upstream SPECC1L (sperm antigen with calponin homology and coiled-coil domains 1-like) and ADORA2A (adenosine A2a receptor) gene sequence has been identified, but it is thought to be non-coding. [provided by RefSeq, Jun 2013]