We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cytoplasmic and membranous expression in macrophages in several tissues including lung macrophages, Kupffer cells in liver and placental Hofbauer cells.
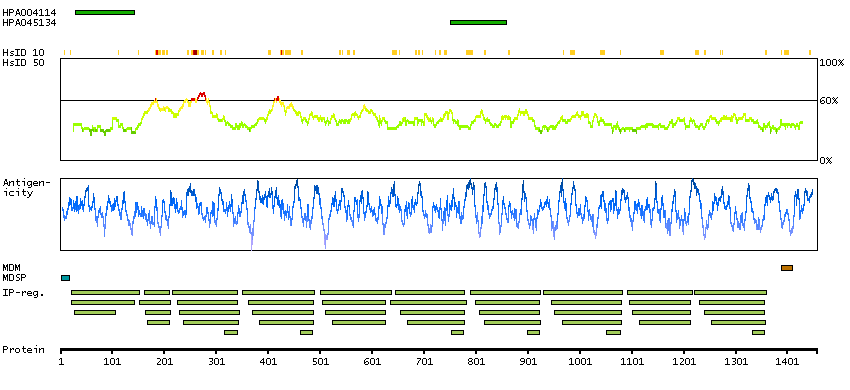
DATA RELIABILITY
Data reliability description
Antibody staining mainly consistent with RNA expression data. Antibody staining in cells/structures not annotated, view images.
The recognition of complex carbohydrate structures on glycoproteins is an important part of several biological processes, including cell-cell recognition, serum glycoprotein turnover, and neutralization of pathogens. The protein encoded by this gene is a type I membrane receptor that mediates the endocytosis of glycoproteins by macrophages. The protein has been shown to bind high-mannose structures on the surface of potentially pathogenic viruses, bacteria, and fungi so that they can be neutralized by phagocytic engulfment.[provided by RefSeq, Sep 2015]