We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
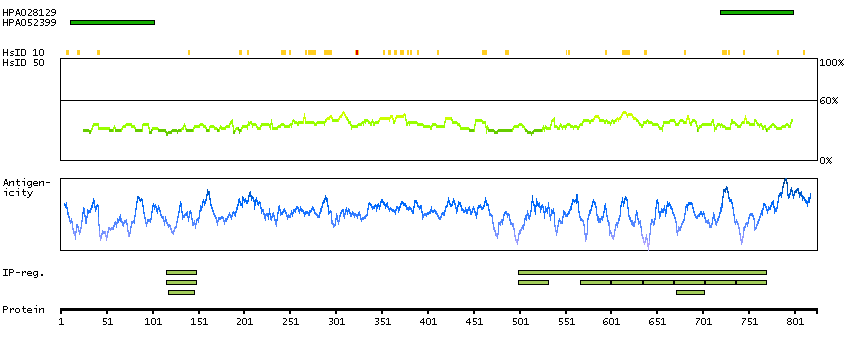
The protein encoded by this gene shares strong similarity with Saccharomyces cerevisiae protein Cdc27, and the gene product of Schizosaccharomyces pombe nuc 2. This protein is a component of the anaphase-promoting complex (APC), which is composed of eight protein subunits and is highly conserved in eukaryotic cells. This complex catalyzes the formation of cyclin B-ubiquitin conjugate, which is responsible for the ubiquitin-mediated proteolysis of B-type cyclins. The protein encoded by this gene and three other members of the APC complex contain tetratricopeptide (TPR) repeats, which are important for protein-protein interactions. This protein was shown to interact with mitotic checkpoint proteins including Mad2, p55CDC and BUBR1, and it may thus be involved in controlling the timing of mitosis. Alternative splicing of this gene results in multiple transcript variants. Related pseudogenes have been identified on chromosomes 2, 22 and Y. [provided by RefSeq, May 2014]
P30260 [Direct mapping] Cell division cycle protein 27 homolog
Show all
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
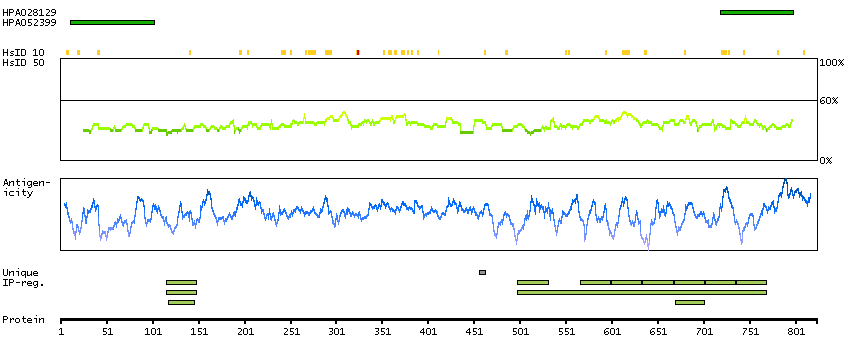
P30260 [Direct mapping] Cell division cycle protein 27 homolog
Show all
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)