We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
In vertebrates, the genes encoding the class of transcription factors called homeobox genes are found in clusters named A, B, C, and D on four separate chromosomes. Expression of these proteins is spatially and temporally regulated during embryonic development. This gene is part of the A cluster on chromosome 7 and encodes a DNA-binding transcription factor which may regulate gene expression, morphogenesis, and differentiation. This gene is involved in the regulation of uterine development and is required for female fertility. Mutations in this gene can cause radio-ulnar synostosis with amegakaryocytic thrombocytopenia. [provided by RefSeq, Jul 2008]
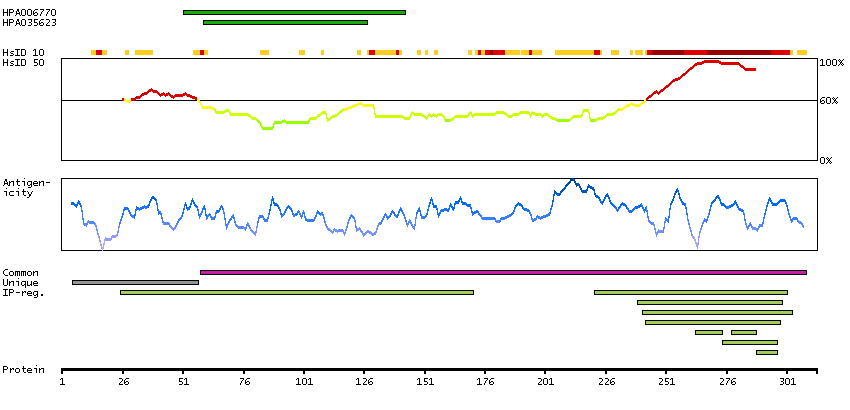
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Ezkurdia et al 2014)
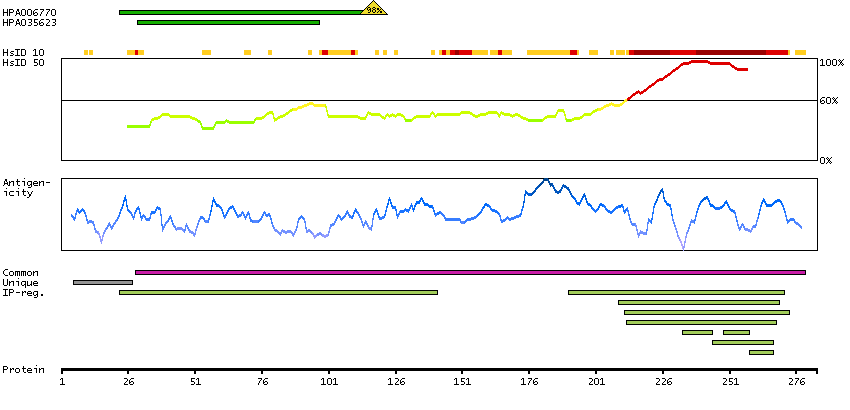
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)