We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
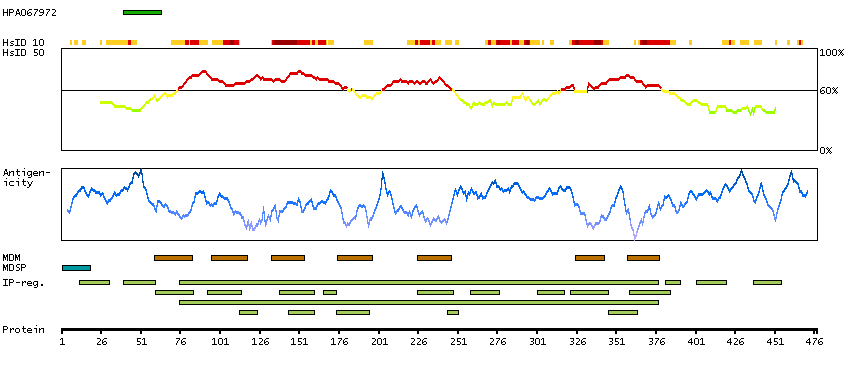
The adrenergic receptors (subtypes alpha 1, alpha 2, beta 1, and beta 2) are a prototypic family of guanine nucleotide binding regulatory protein-coupled receptors that mediate the physiological effects of the hormone epinephrine and the neurotransmitter norepinephrine. Specific polymorphisms in this gene have been shown to affect the resting heart rate and can be involved in heart failure. [provided by RefSeq, Jul 2008]