We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
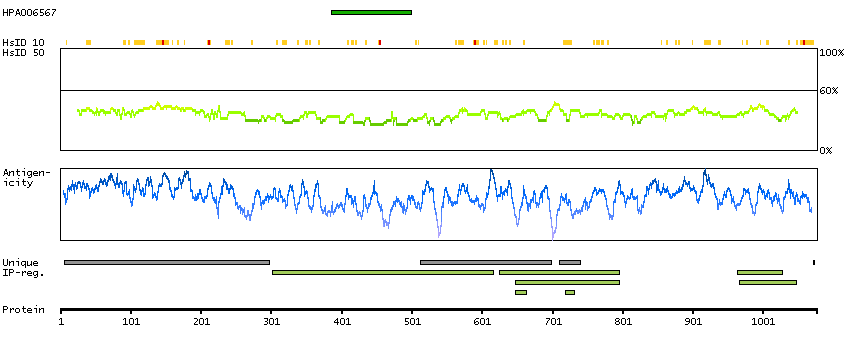
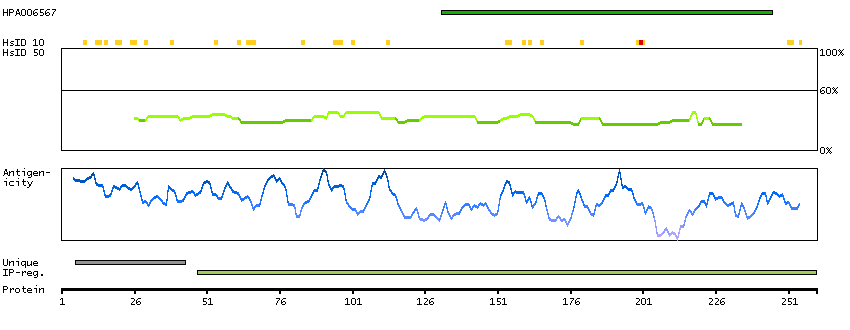
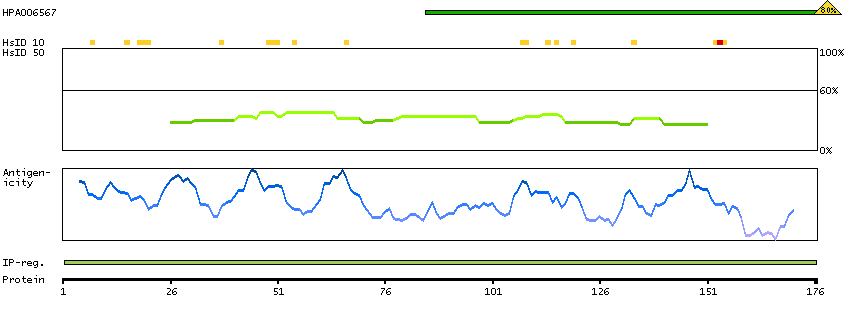
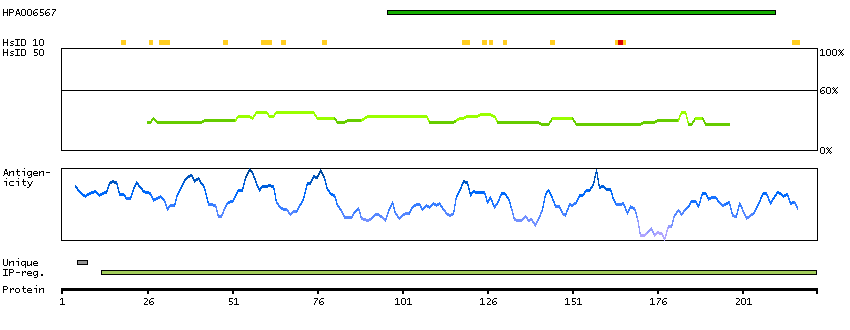
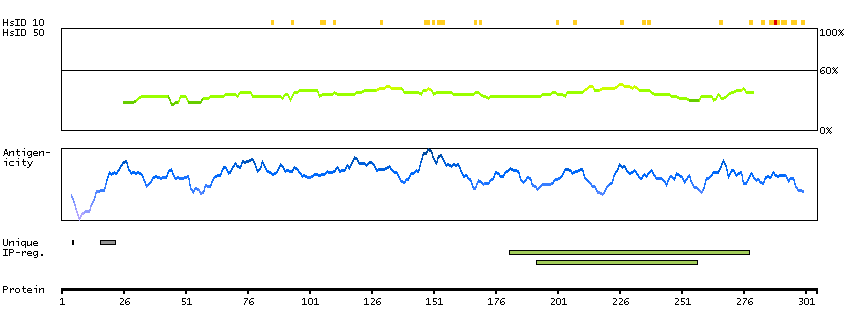
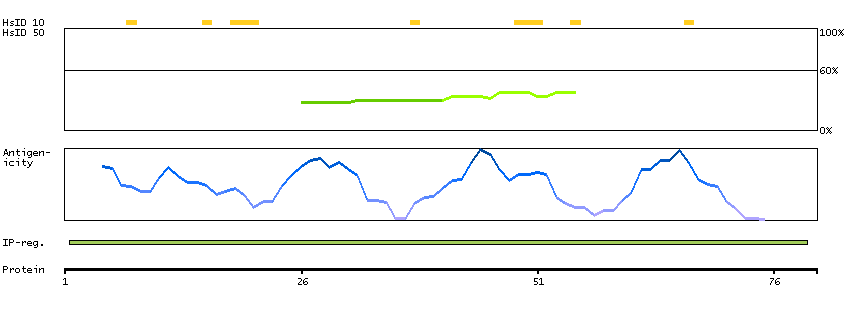
The protein encoded by this gene has a long and a short form, generated by use of alternative translational start codons. The long form is expressed in steroidogenic tissues such as testis, where it converts cholesteryl esters to free cholesterol for steroid hormone production. The short form is expressed in adipose tissue, among others, where it hydrolyzes stored triglycerides to free fatty acids. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Hydrolases Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)