We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
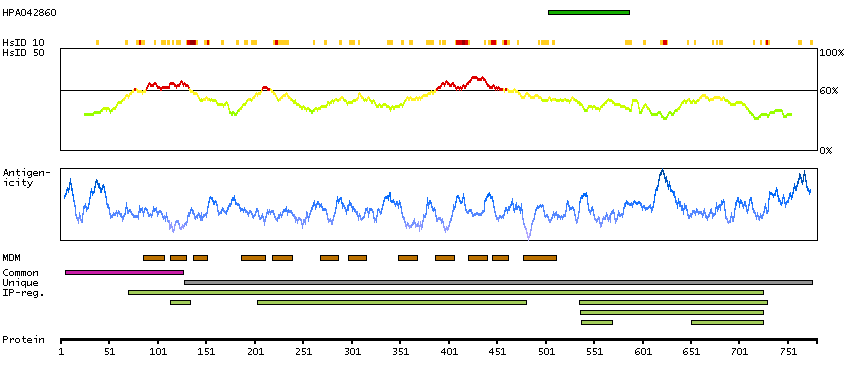
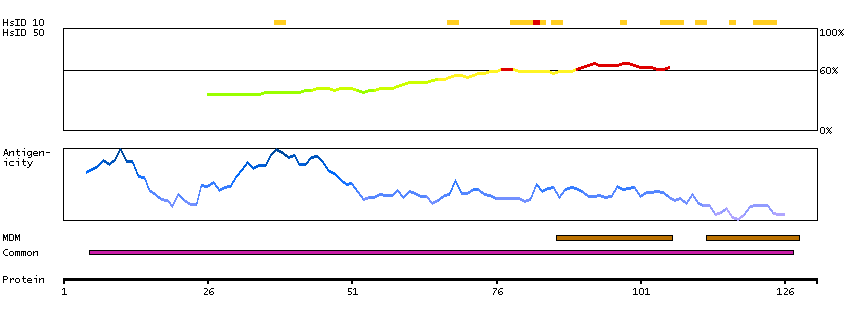
Solute carrier family 26 (anion exchanger), member 4 (HGNC Symbol)
Entrez gene summary
Mutations in this gene are associated with Pendred syndrome, the most common form of syndromic deafness, an autosomal-recessive disease. It is highly homologous to the SLC26A3 gene; they have similar genomic structures and this gene is located 3' of the SLC26A3 gene. The encoded protein has homology to sulfate transporters. [provided by RefSeq, Jul 2008]