We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
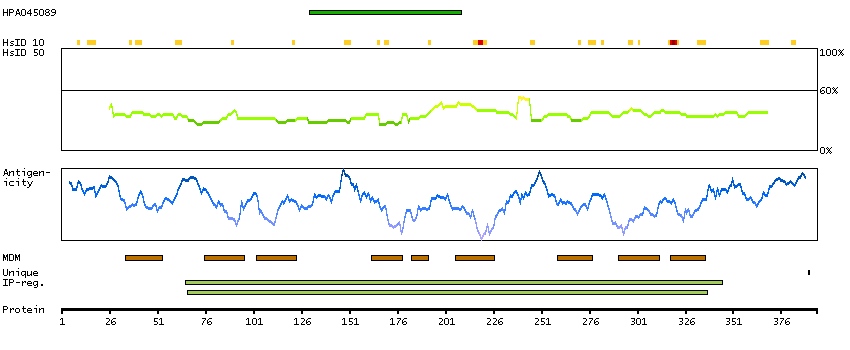
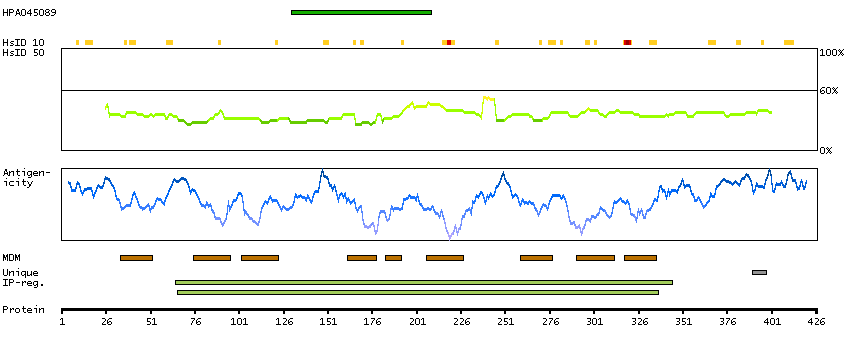
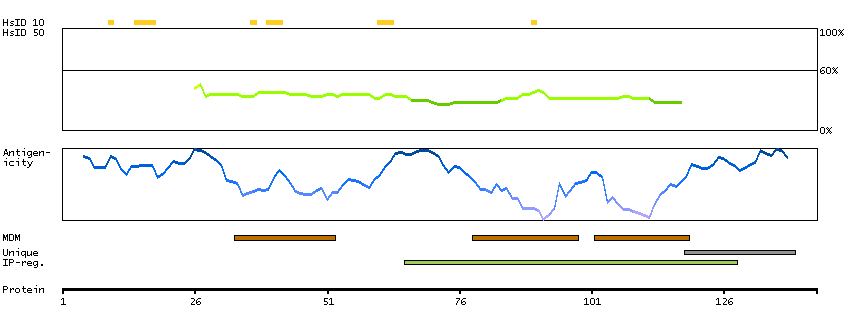
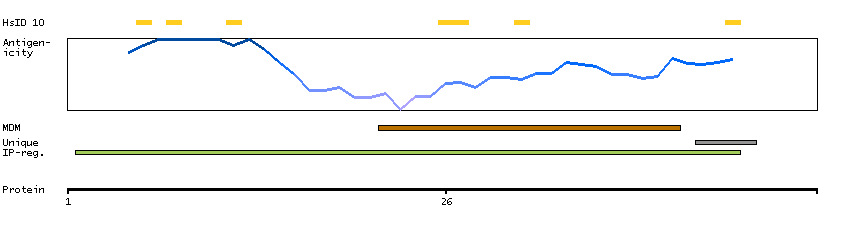
The protein encoded by this gene, which localizes to the endoplasmic reticulum, catalyzes intramembrane proteolysis of some signal peptides after they have been cleaved from a preprotein. This activity is required to generate signal sequence-derived human lymphocyte antigen-E epitopes that are recognized by the immune system, and to process hepatitis C virus core protein. The encoded protein is an integral membrane protein with sequence motifs characteristic of the presenilin-type aspartic proteases. Multiple transcript variants encoding several different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]