We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
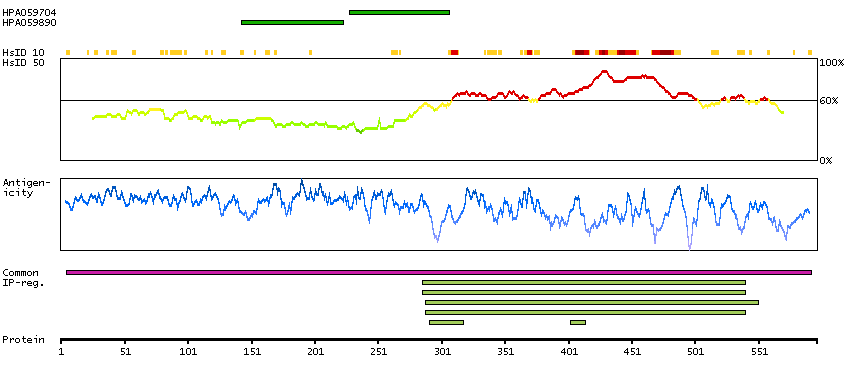
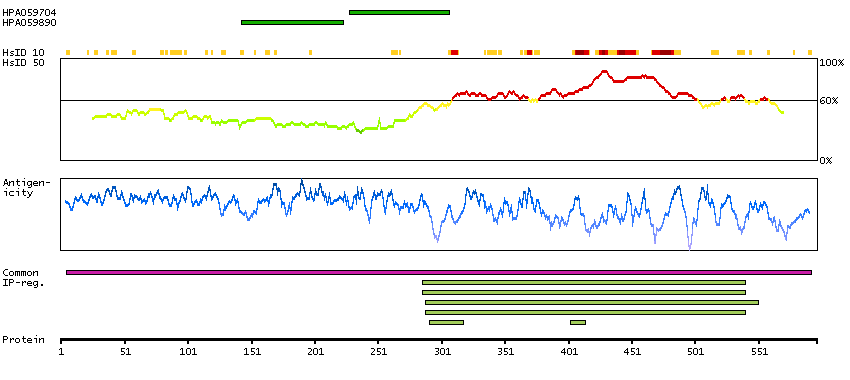
This gene encodes a myosin light chain kinase, a calcium/calmodulin dependent enzyme, that is exclusively expressed in adult skeletal muscle. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Transferases Kinases CAMK Ser/Thr protein kinases MEMSAT-SVM predicted membrane proteins SCAMPI predicted membrane proteins Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)