We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
C-type lectin domain family 4, member M (HGNC Symbol)
Entrez gene summary
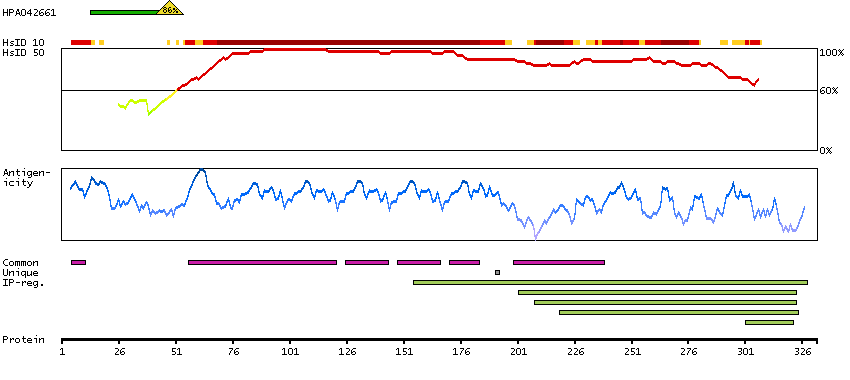
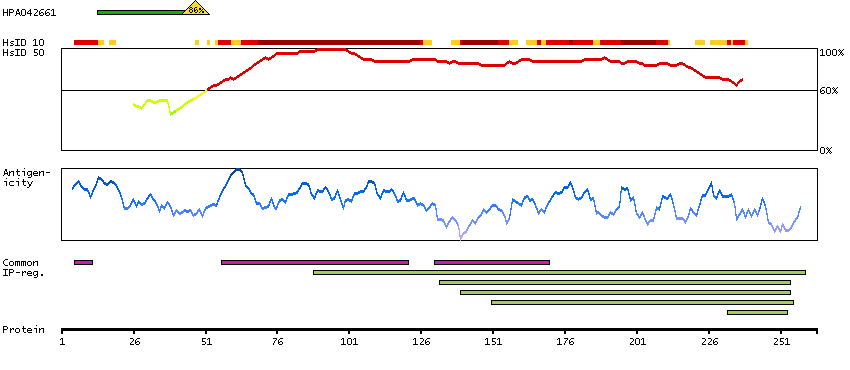
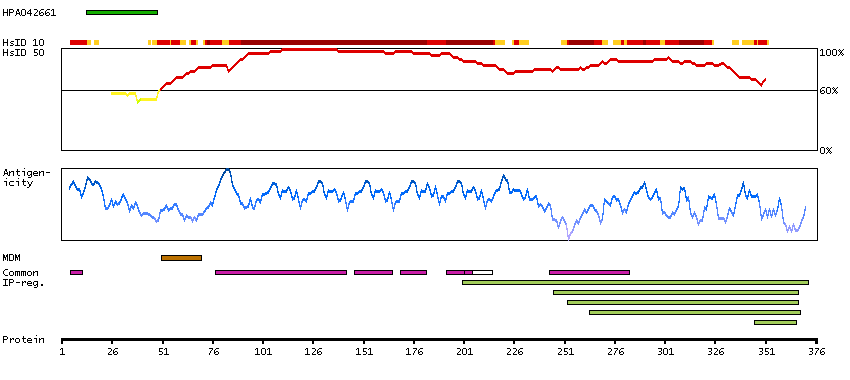
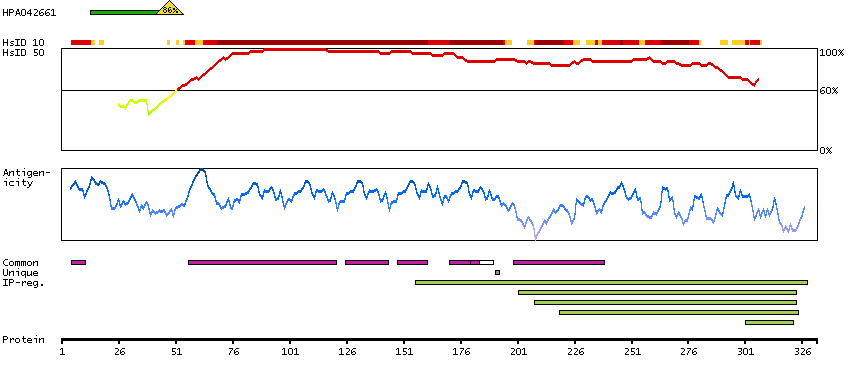
This gene encodes a transmembrane receptor and is often referred to as L-SIGN because of its expression in the endothelial cells of the lymph nodes and liver. The encoded protein is involved in the innate immune system and recognizes numerous evolutionarily divergent pathogens ranging from parasites to viruses, with a large impact on public health. The protein is organized into three distinct domains: an N-terminal transmembrane domain, a tandem-repeat neck domain and C-type lectin carbohydrate recognition domain. The extracellular region consisting of the C-type lectin and neck domains has a dual function as a pathogen recognition receptor and a cell adhesion receptor by binding carbohydrate ligands on the surface of microbes and endogenous cells. The neck region is important for homo-oligomerization which allows the receptor to bind multivalent ligands with high avidity. Variations in the number of 23 amino acid repeats in the neck domain of this protein are common and have a significant impact on ligand binding ability. This gene is closely related in terms of both sequence and function to a neighboring gene (GeneID 30835; often referred to as DC-SIGN or CD209). DC-SIGN and L-SIGN differ in their ligand-binding properties and distribution. Alternative splicing results in multiple variants.[provided by RefSeq, Feb 2009]