We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
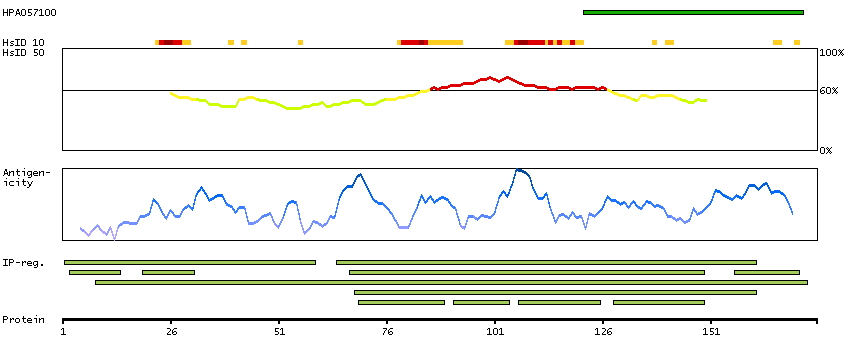
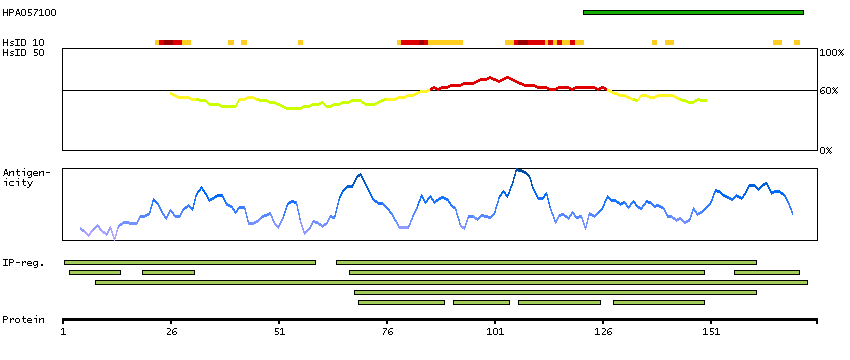
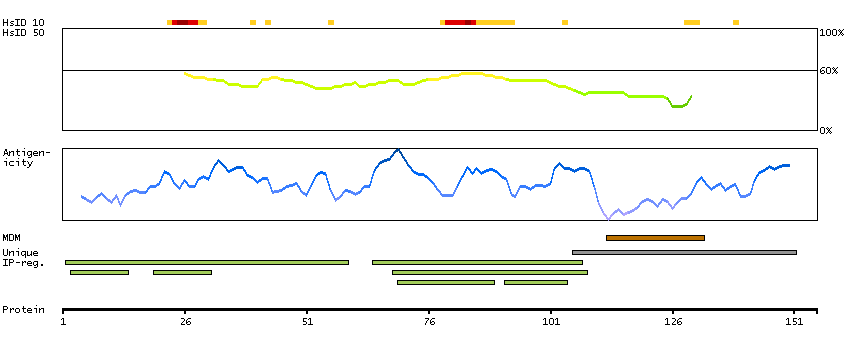
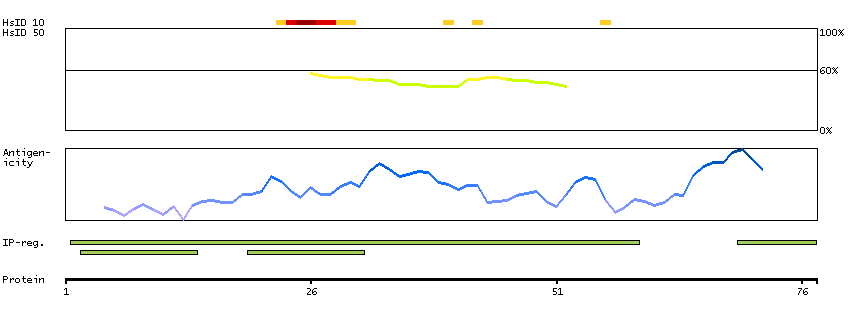
Mammalian lens crystallins are divided into alpha, beta, and gamma families. Alpha crystallins are composed of two gene products: alpha-A and alpha-B, for acidic and basic, respectively. Alpha crystallins can be induced by heat shock and are members of the small heat shock protein (HSP20) family. They act as molecular chaperones although they do not renature proteins and release them in the fashion of a true chaperone; instead they hold them in large soluble aggregates. Post-translational modifications decrease the ability to chaperone. These heterogeneous aggregates consist of 30-40 subunits; the alpha-A and alpha-B subunits have a 3:1 ratio, respectively. Two additional functions of alpha crystallins are an autokinase activity and participation in the intracellular architecture. The encoded protein has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions. Alpha-A and alpha-B gene products are differentially expressed; alpha-A is preferentially restricted to the lens and alpha-B is expressed widely in many tissues and organs. Elevated expression of alpha-B crystallin occurs in many neurological diseases; a missense mutation cosegregated in a family with a desmin-related myopathy. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2014]
P02511 [Direct mapping] Alpha-crystallin B chain V9HW27 [Target identity:100%; Query identity:100%] Crystallin, alpha B, isoform CRA_a; Epididymis secretory protein Li 101
Show all
Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)