We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Selective cytoplasmic expression in heart and skeletal muscle.
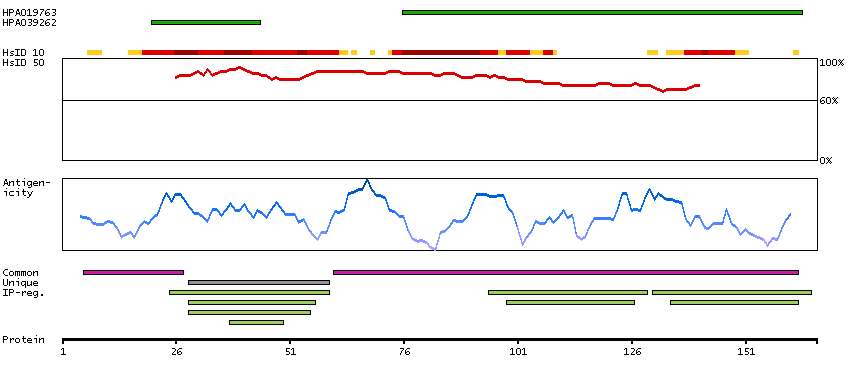
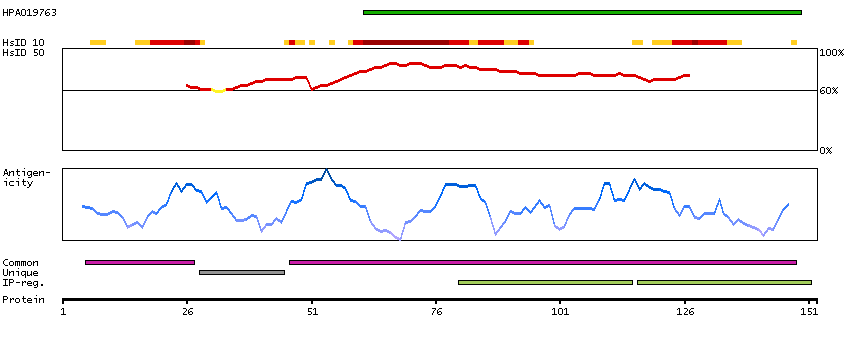
DATA RELIABILITY
Data reliability description
Antibody staining mainly consistent with RNA expression data. Caution, targets protein from more than one gene. Presumed off target binding observed and disregarded.
Thus gene encodes the regulatory light chain associated with cardiac myosin beta (or slow) heavy chain. Ca+ triggers the phosphorylation of regulatory light chain that in turn triggers contraction. Mutations in this gene are associated with mid-left ventricular chamber type hypertrophic cardiomyopathy. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)