We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Group enriched (hippocampus, brain, caudate, cerebellum, pituitary gland)
Protein evidence
Evidence at transcript level
Protein expression
Cytoplasmic expression in several tissues.
DATA RELIABILITY
Data reliability description
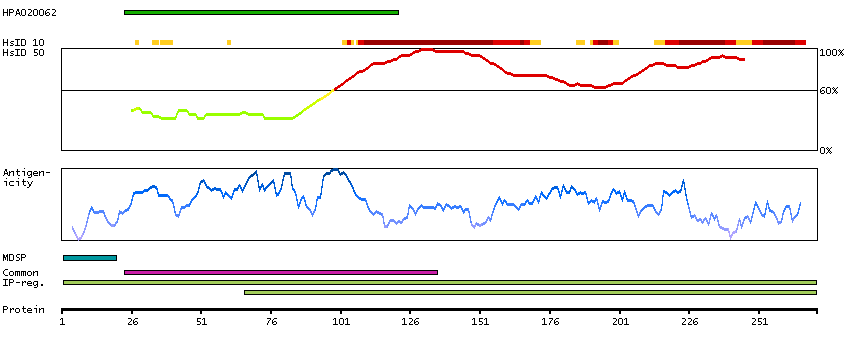
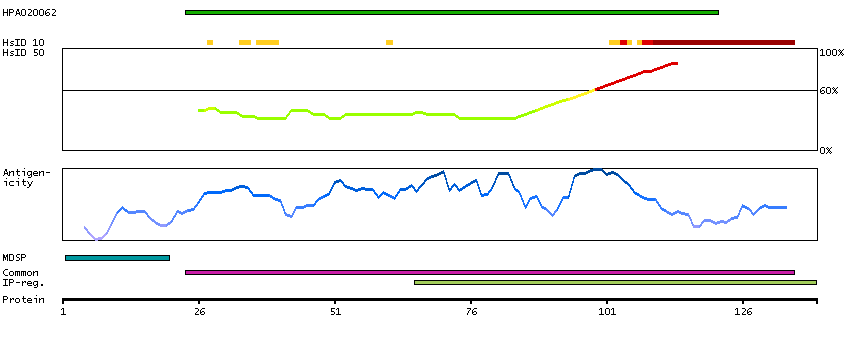
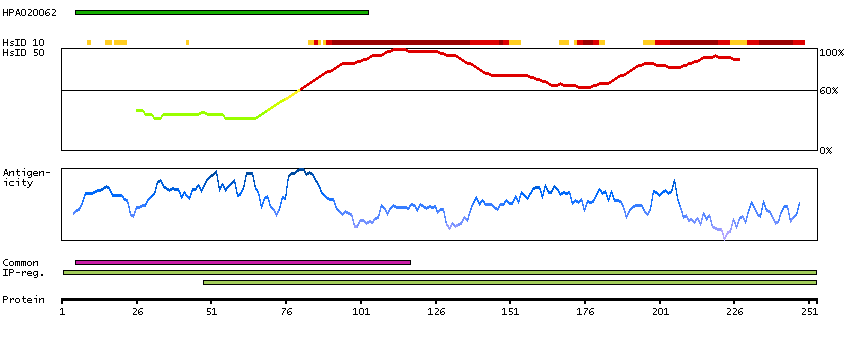
Antibody staining mainly not consistent with RNA expression data. Secreted protein, tissue location of RNA and protein might differ and correlation is complex.
This gene is a member of the neurexophilin family and encodes a secreted protein with a variable N-terminal domain, a highly conserved, N-glycosylated central domain, a short linker region, and a cysteine-rich C-terminal domain. This protein forms a very tight complex with alpha neurexins, a group of proteins that promote adhesion between dendrites and axons. [provided by RefSeq, Jul 2008]