We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
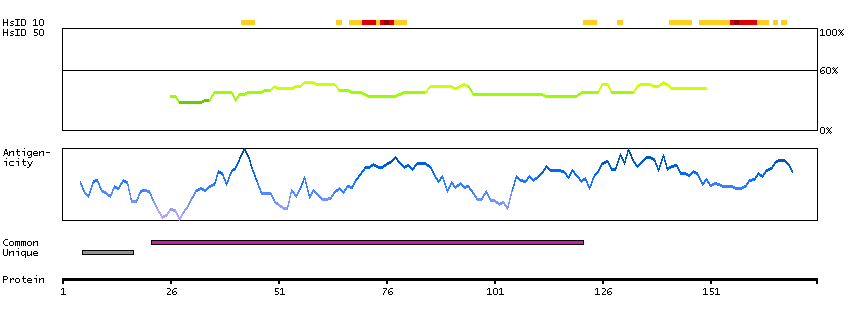
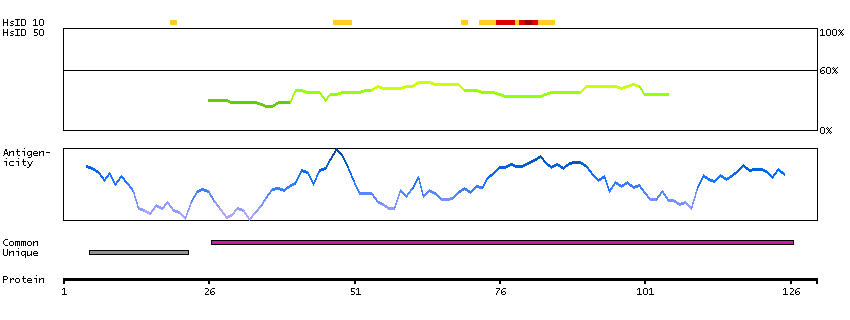
This gene is upstream of, and in a head-to-head orientation with the gene for the mitochondrial ribosomal protein L34. The predicted protein contains alpha/beta hydrolase fold and secretory lipase domains. [provided by RefSeq, Jul 2008]