We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
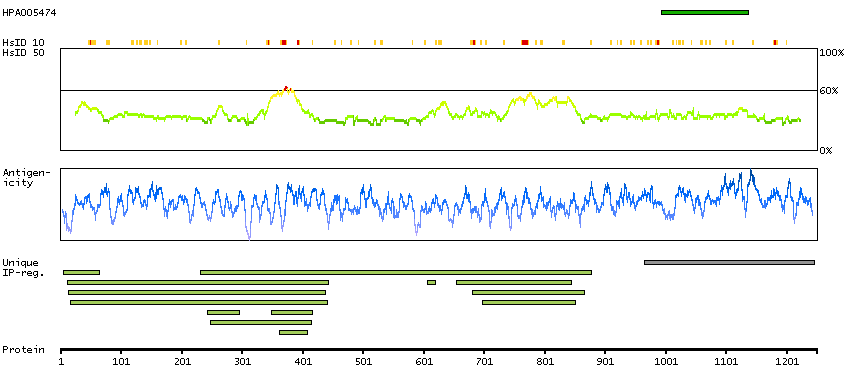
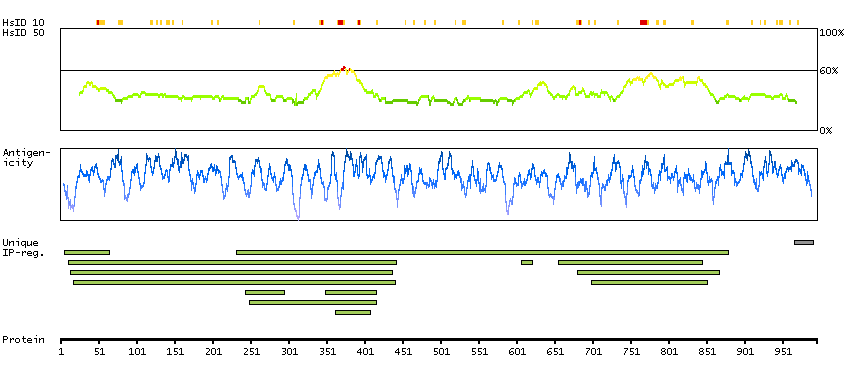
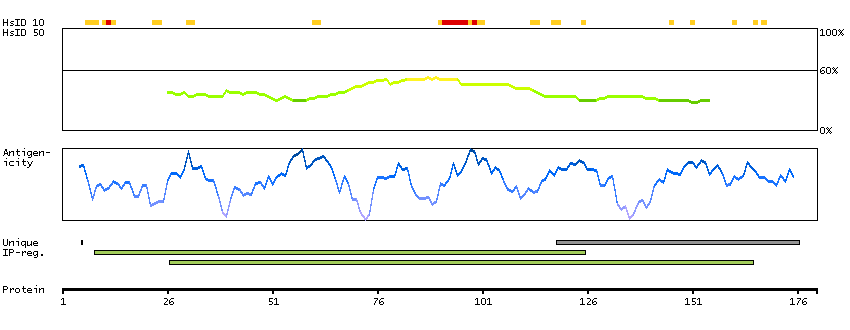
BRCA1 interacting protein C-terminal helicase 1 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the RecQ DEAH helicase family and interacts with the BRCT repeats of breast cancer, type 1 (BRCA1). The bound complex is important in the normal double-strand break repair function of breast cancer, type 1 (BRCA1). This gene may be a target of germline cancer-inducing mutations. [provided by RefSeq, Jul 2008]
Q9BX63 [Direct mapping] Fanconi anemia group J protein A0A024QZ45 [Target identity:100%; Query identity:100%] BRCA1 interacting protein C-terminal helicase 1, isoform CRA_a
Show all
Enzymes ENZYME proteins Hydrolases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Germline Mutations COSMIC Frameshift Mutations Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)