We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
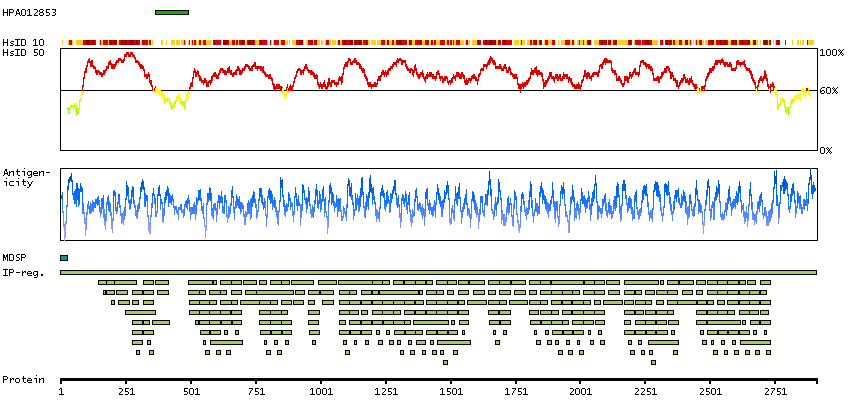
Selective expression in placental trophoblasts as well as extracellular fibers and basement membranes in several tissues including in placenta and testis.
DATA RELIABILITY
Data reliability description
Antibody staining consistent with RNA expression data.
The protein encoded by this gene is a component of connective tissue microfibrils and may be involved in elastic fiber assembly. Mutations in this gene cause congenital contractural arachnodactyly. [provided by RefSeq, Jul 2008]