We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
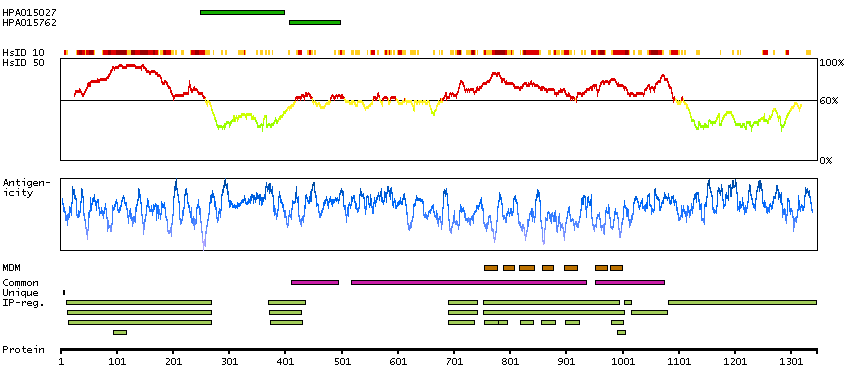
Adhesion G protein-coupled receptor L3 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the latrophilin subfamily of G-protein coupled receptors (GPCR). Latrophilins may function in both cell adhesion and signal transduction. In experiments with non-human species, endogenous proteolytic cleavage within a cysteine-rich GPS (G-protein-coupled-receptor proteolysis site) domain resulted in two subunits (a large extracellular N-terminal cell adhesion subunit and a subunit with substantial similarity to the secretin/calcitonin family of GPCRs) being non-covalently bound at the cell membrane. [provided by RefSeq, Jul 2008]