We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
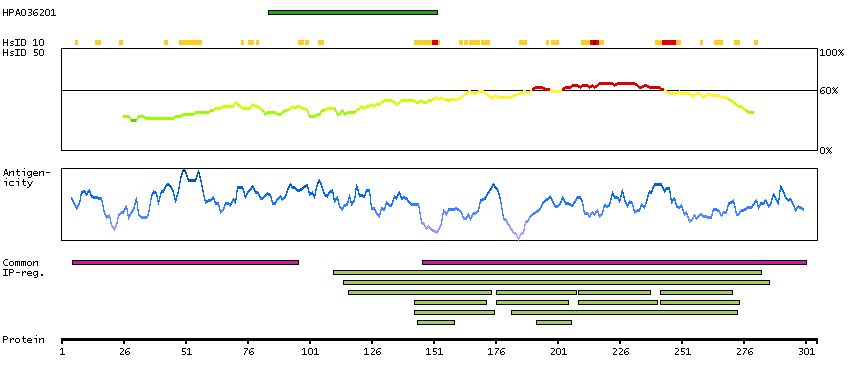
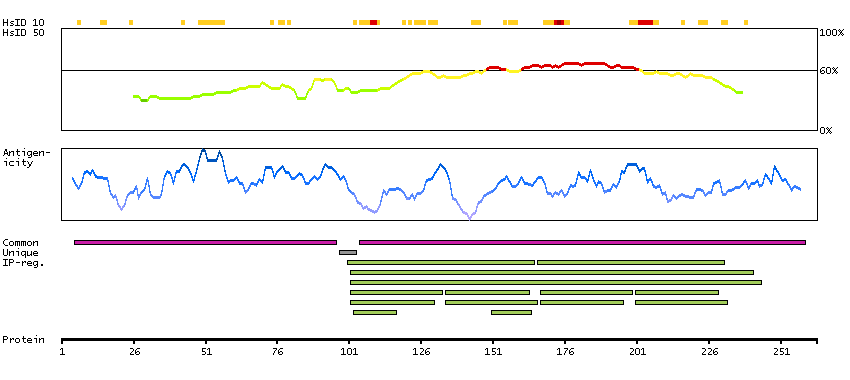
This gene is a member of the muscle ankyrin repeat protein (MARP) family and encodes a protein with four tandem ankyrin-like repeats. The protein is localized to the nucleus, functioning as a transcriptional regulator. Expression of this protein is induced during recovery following starvation. [provided by RefSeq, Jul 2008]