We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Component C2 is a serum glycoprotein that functions as part of the classical pathway of the complement system. Activated C1 cleaves C2 into C2a and C2b. The serine proteinase C2a then combines with complement factor 4b to create the C3 or C5 convertase. Deficiency of C2 has been reported to associated with certain autoimmune diseases and SNPs in this gene have been associated with altered susceptibility to age-related macular degeneration. This gene localizes within the class III region of the MHC on the short arm of chromosome 6. Alternative splicing results in multiple transcript variants encoding distinct isoforms. Additional transcript variants have been described in publications but their full-length sequence has not been determined.[provided by RefSeq, Mar 2009]
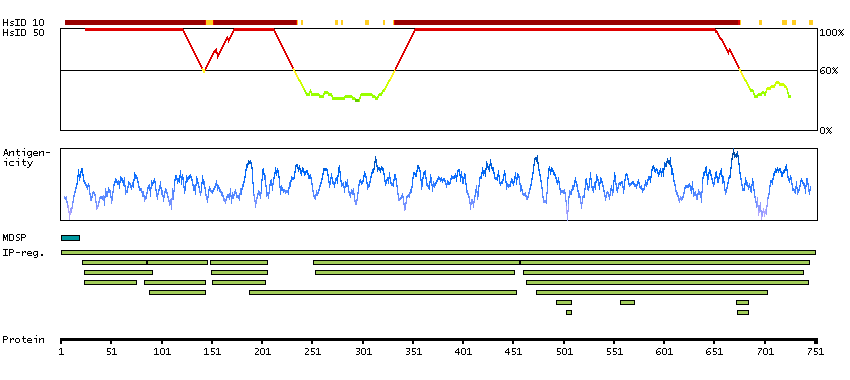
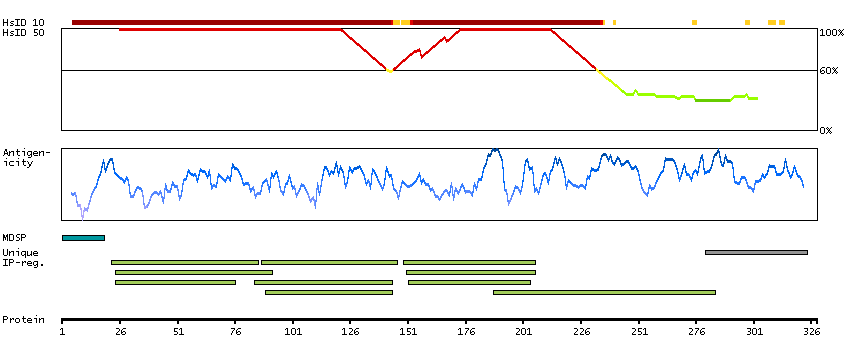
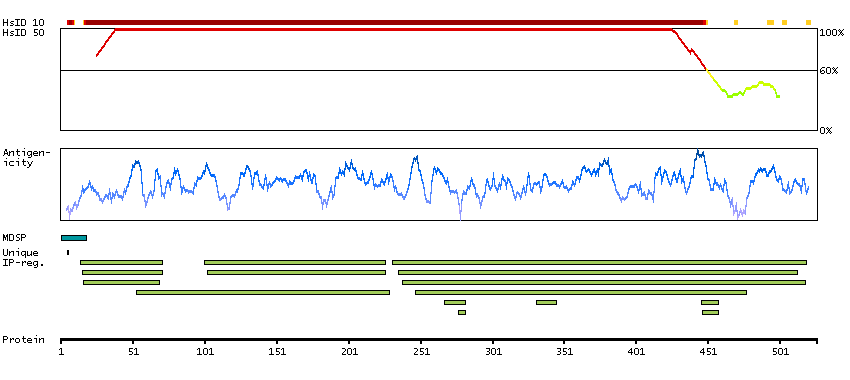
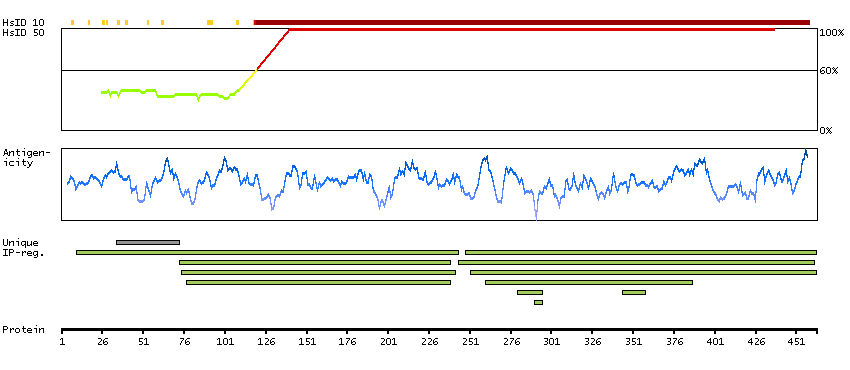
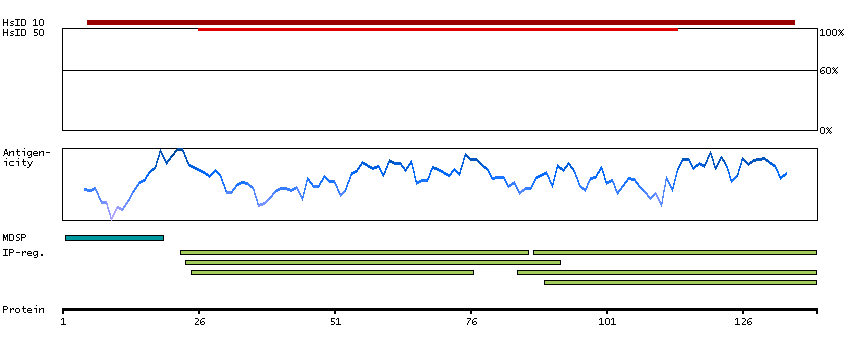
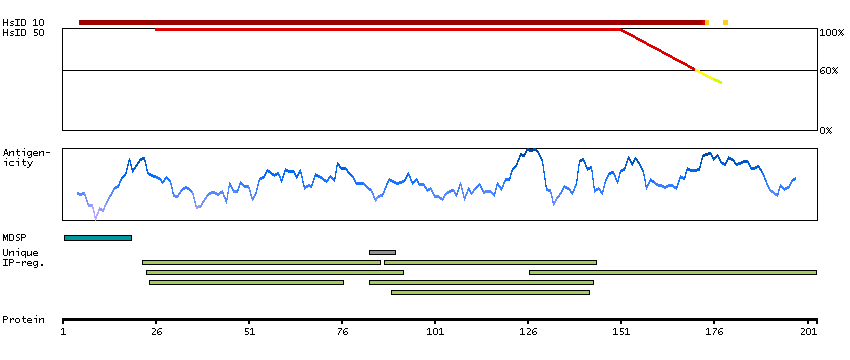
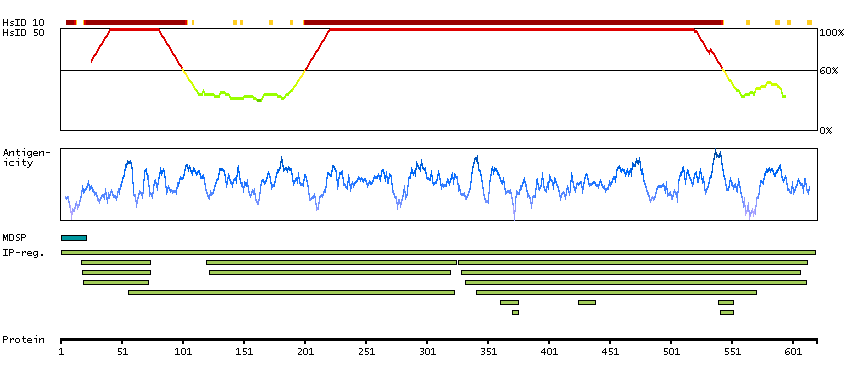
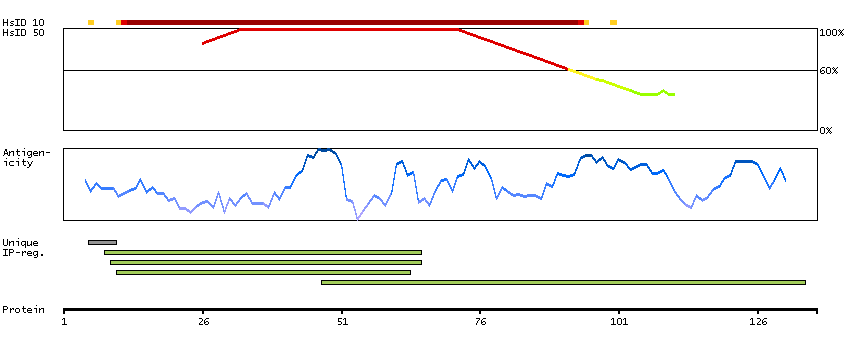
P06681 [Direct mapping] Complement C2 Complement C2b fragment Complement C2a fragment
Show all
Enzymes ENZYME proteins Hydrolases Peptidases Serine-type peptidases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)