We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
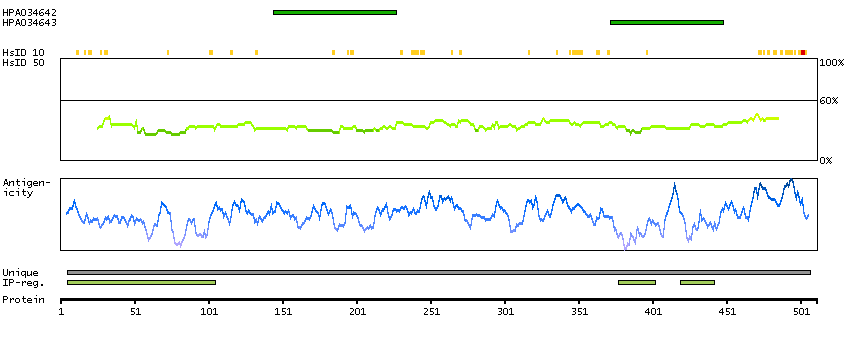
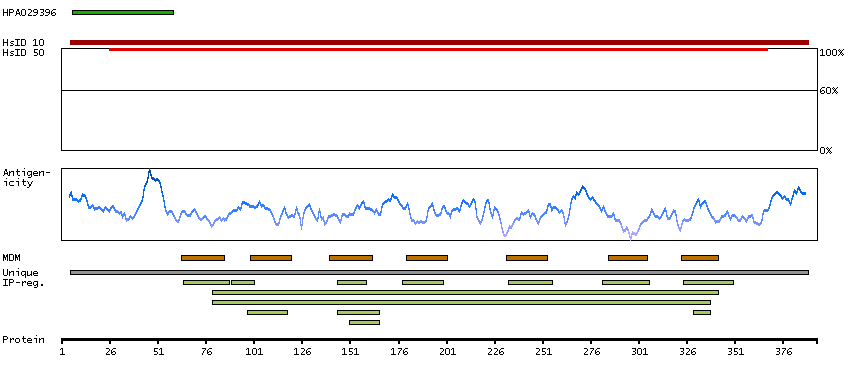
C2ORF13 is a component of the cellular response to chromosomal DNA single- and double-strand breaks (Iles et al., 2007 [PubMed 17353262]).[supplied by OMIM, Mar 2008]