We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (skin, urinary bladder, vagina, esophagus)
FANTOM5 dataset
Organ
Expression
Alphabetical
RNA tissue category: Tissue enhanced (tonsil)
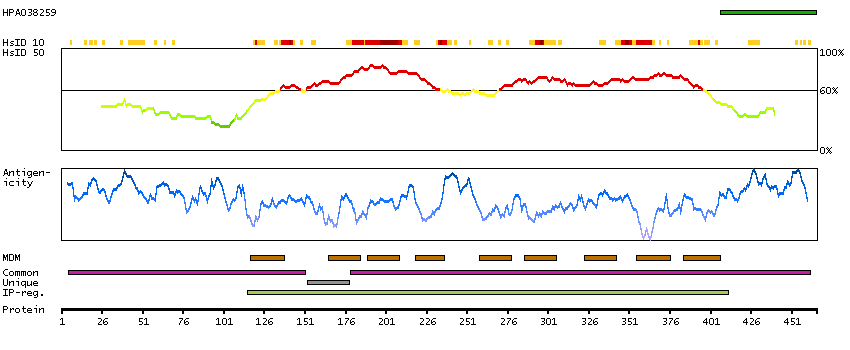
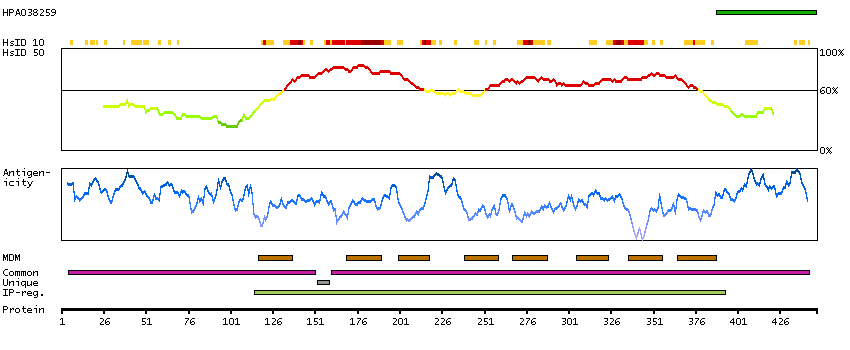
GENE INFORMATION
Gene name
NIPAL4 (HGNC Symbol)
Synonyms
ICHYN
Description
NIPA-like domain containing 4 (HGNC Symbol)
Entrez gene summary
This gene likely encodes a membrane receptor. Mutations in this gene have been associated with autosomal recessive congenital ichthyosis. [provided by RefSeq, Feb 2010]
GO:0015095 [magnesium ion transmembrane transporter activity] GO:0015693 [magnesium ion transport] GO:0016021 [integral component of membrane] GO:1903830 [magnesium ion transmembrane transport]
GO:0015095 [magnesium ion transmembrane transporter activity] GO:0015693 [magnesium ion transport] GO:0016021 [integral component of membrane] GO:1903830 [magnesium ion transmembrane transport]