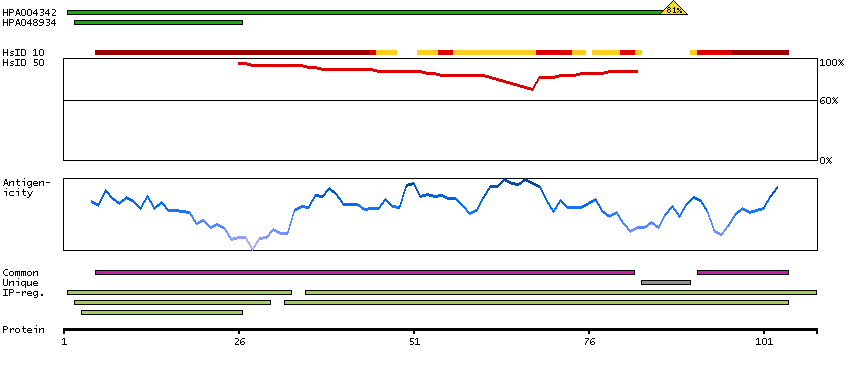
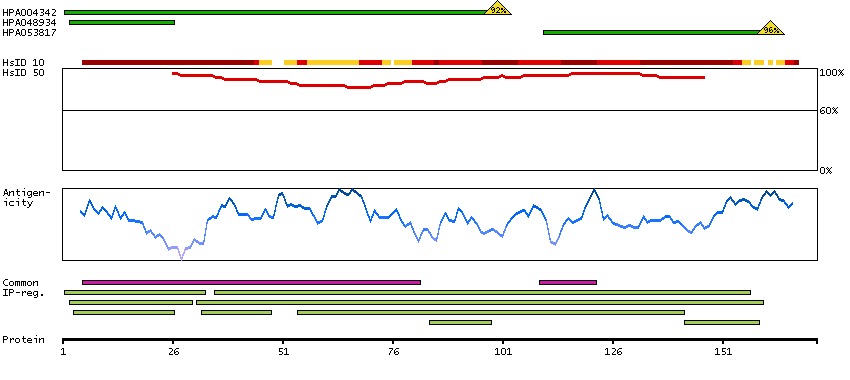
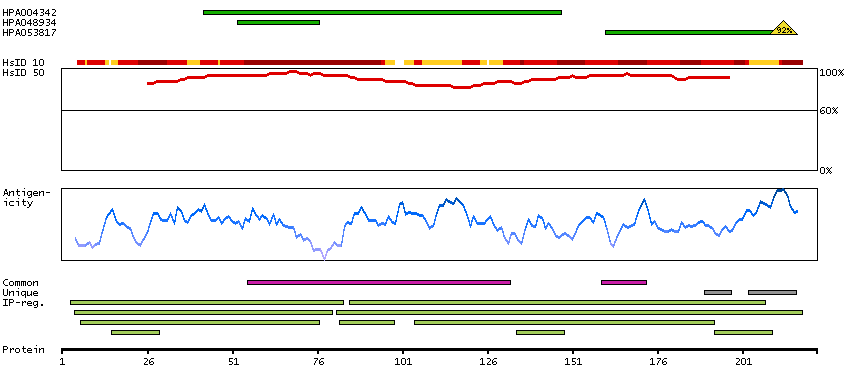
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cytosolic and membrane-bound forms of glutathione S-transferase are encoded by two distinct supergene families. These enzymes are involved in cellular defense against toxic, carcinogenic, and pharmacologically active electrophilic compounds. At present, eight distinct classes of the soluble cytoplasmic mammalian glutathione S-transferases have been identified: alpha, kappa, mu, omega, pi, sigma, theta and zeta. This gene encodes a glutathione S-tranferase belonging to the alpha class genes that are located in a cluster mapped to chromosome 6. Genes of the alpha class are highly related and encode enzymes with glutathione peroxidase activity. However, during evolution, this alpha class gene diverged accumulating mutations in the active site that resulted in differences in substrate specificity and catalytic activity. The enzyme encoded by this gene catalyzes the double bond isomerization of precursors for progesterone and testosterone during the biosynthesis of steroid hormones. An additional transcript variant has been identified, but its full length sequence has not been determined. [provided by RefSeq, Jul 2008]