We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
ATPase, Ca++ transporting, cardiac muscle, fast twitch 1 (HGNC Symbol)
Entrez gene summary
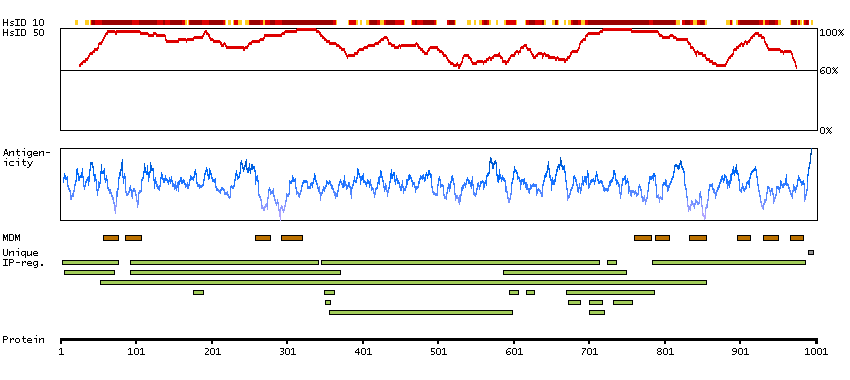
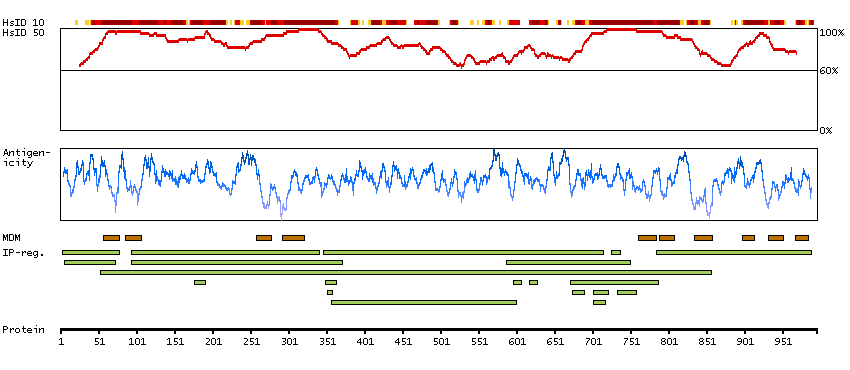
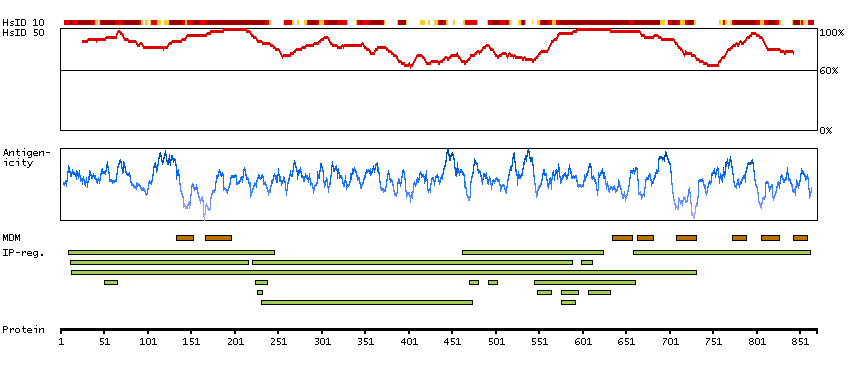
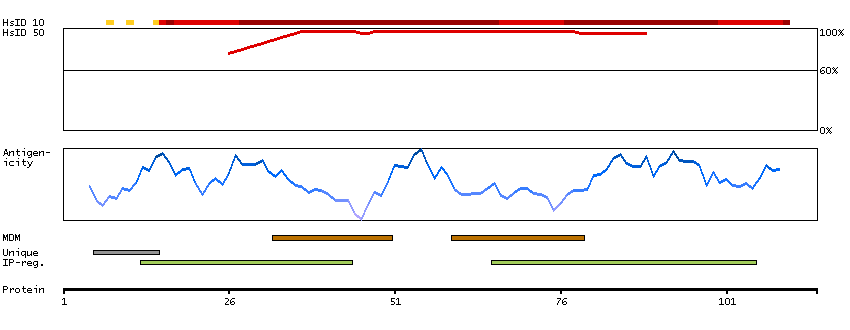
This gene encodes one of the SERCA Ca(2+)-ATPases, which are intracellular pumps located in the sarcoplasmic or endoplasmic reticula of muscle cells. This enzyme catalyzes the hydrolysis of ATP coupled with the translocation of calcium from the cytosol to the sarcoplasmic reticulum lumen, and is involved in muscular excitation and contraction. Mutations in this gene cause some autosomal recessive forms of Brody disease, characterized by increasing impairment of muscular relaxation during exercise. Alternative splicing results in three transcript variants encoding different isoforms. [provided by RefSeq, Oct 2013]