We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Alcohol dehydrogenase 5 (class III), chi polypeptide (HGNC Symbol)
Entrez gene summary
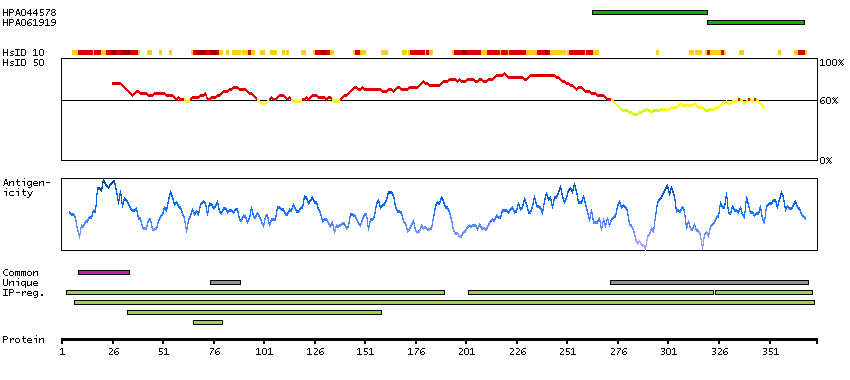
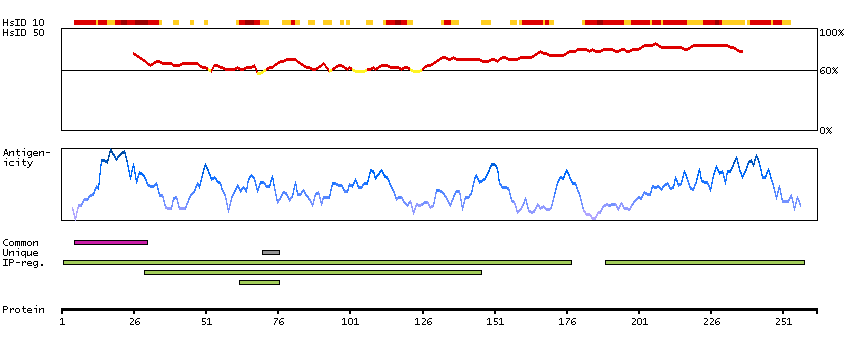
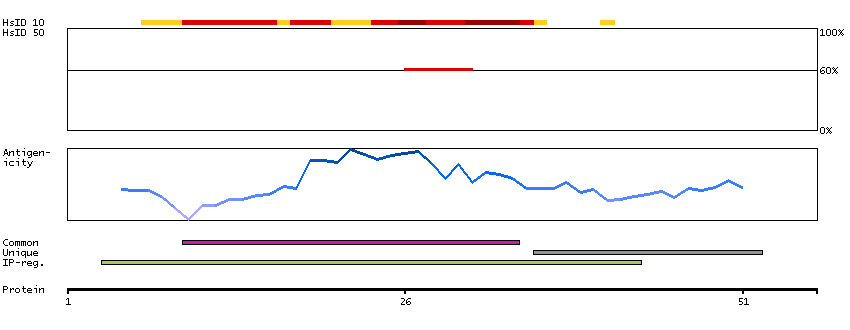
This gene encodes a member of the alcohol dehydrogenase family. Members of this family metabolize a wide variety of substrates, including ethanol, retinol, other aliphatic alcohols, hydroxysteroids, and lipid peroxidation products. The encoded protein forms a homodimer. It has virtually no activity for ethanol oxidation, but exhibits high activity for oxidation of long-chain primary alcohols and for oxidation of S-hydroxymethyl-glutathione, a spontaneous adduct between formaldehyde and glutathione. This enzyme is an important component of cellular metabolism for the elimination of formaldehyde, a potent irritant and sensitizing agent that causes lacrymation, rhinitis, pharyngitis, and contact dermatitis. The human genome contains several non-transcribed pseudogenes related to this gene. [provided by RefSeq, Oct 2008]