We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
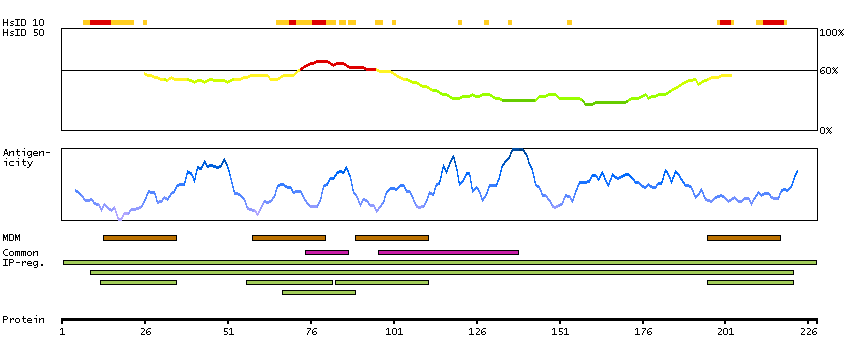
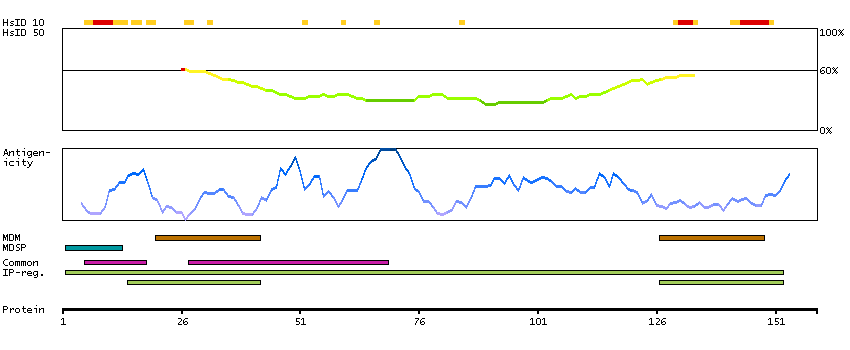
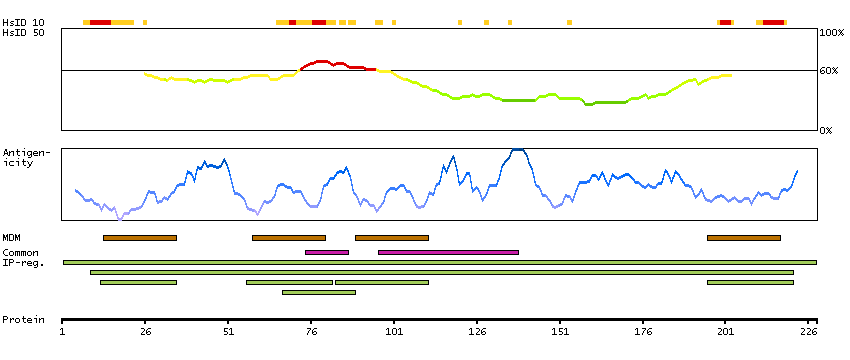
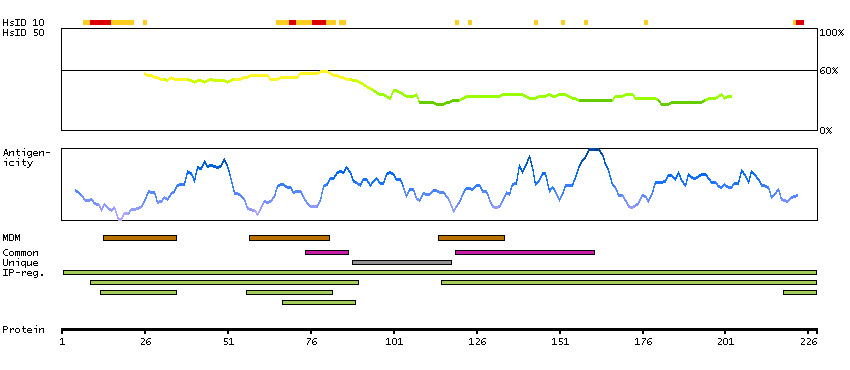
This gene encodes a member of the transmembrane 4 superfamily, also known as the tetraspanin family. Tetraspanins are cell surface glycoproteins with four transmembrane domains that form multimeric complexes with other cell surface proteins. The encoded protein functions in many cellular processes including differentiation, adhesion, and signal transduction, and expression of this gene plays a critical role in the suppression of cancer cell motility and metastasis. [provided by RefSeq, Jan 2011]