We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
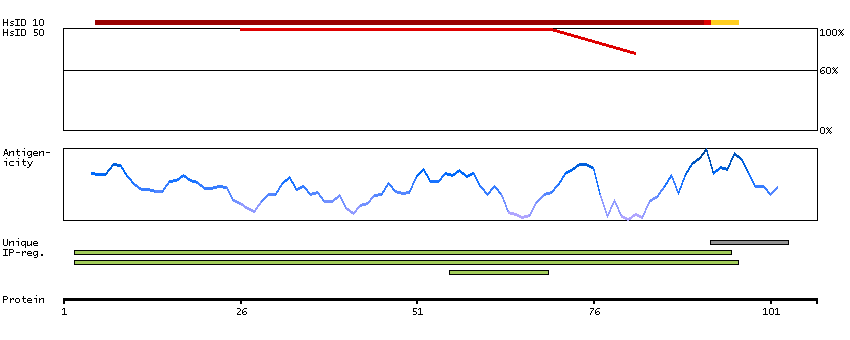
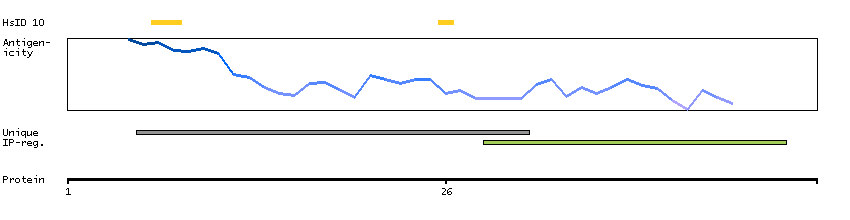
D-dopachrome tautomerase converts D-dopachrome into 5,6-dihydroxyindole. The DDT gene is related to the migration inhibitory factor (MIF) in terms of sequence, enzyme activity, and gene structure. DDT and MIF are closely linked on chromosome 22. [provided by RefSeq, Jul 2008]
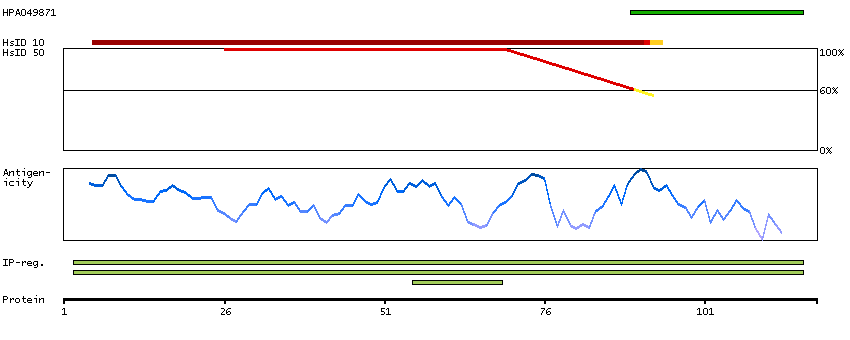
P30046 [Direct mapping] D-dopachrome decarboxylase Q53Y51 [Target identity:100%; Query identity:100%] D-dopachrome tautomerase; D-dopachrome tautomerase 1; D-dopachrome tautomerase, isoform CRA_b; cDNA FLJ76419, highly similar to Homo sapiens D-dopachrome tautomerase (DDT), mRNA
Show all
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
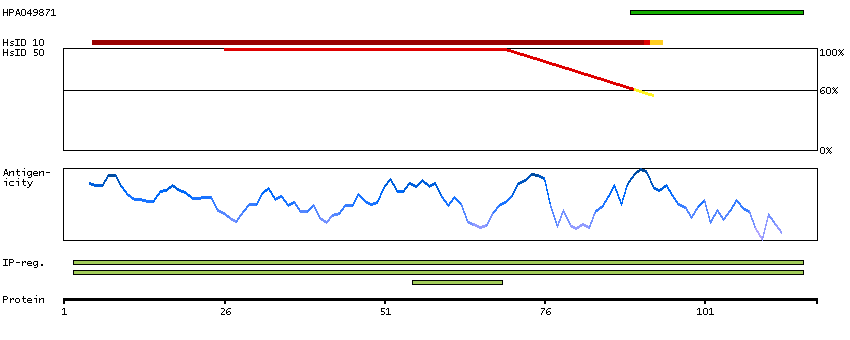
P30046 [Direct mapping] D-dopachrome decarboxylase Q53Y51 [Target identity:100%; Query identity:100%] D-dopachrome tautomerase; D-dopachrome tautomerase 1; D-dopachrome tautomerase, isoform CRA_b; cDNA FLJ76419, highly similar to Homo sapiens D-dopachrome tautomerase (DDT), mRNA
Show all
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)