We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
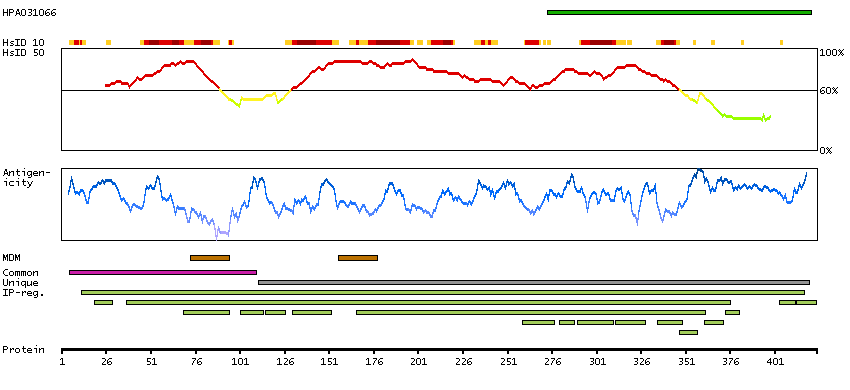
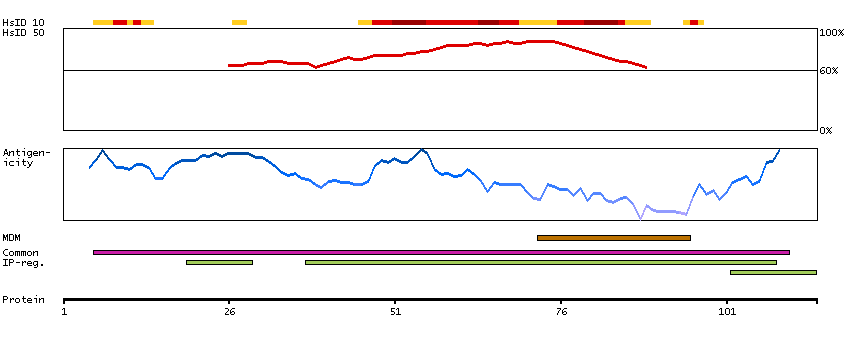
Potassium channel, inwardly rectifying subfamily J, member 8 (HGNC Symbol)
Entrez gene summary
Potassium channels are present in most mammalian cells, where they participate in a wide range of physiologic responses. The protein encoded by this gene is an integral membrane protein and inward-rectifier type potassium channel. The encoded protein, which has a greater tendency to allow potassium to flow into a cell rather than out of a cell, is controlled by G-proteins. Defects in this gene may be a cause of J-wave syndromes and sudden infant death syndrome (SIDS). [provided by RefSeq, May 2012]