We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Interferon (alpha, beta and omega) receptor 1 (HGNC Symbol)
Entrez gene summary
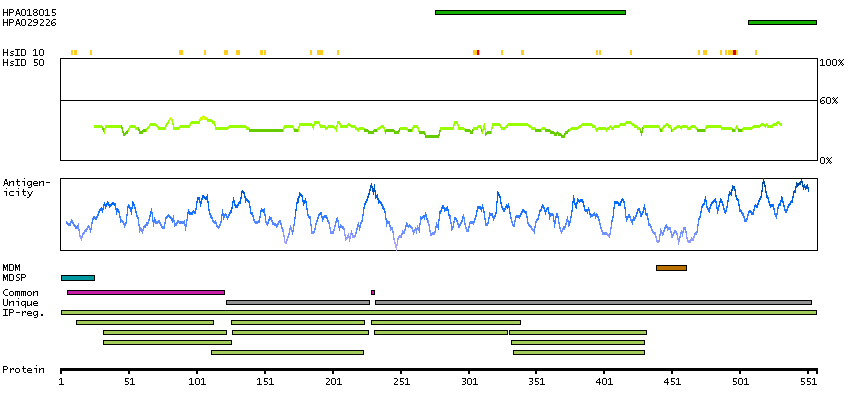
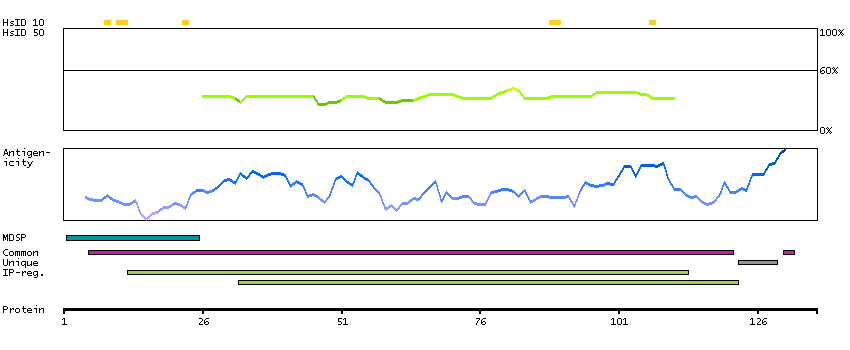
The protein encoded by this gene is a type I membrane protein that forms one of the two chains of a receptor for interferons alpha and beta. Binding and activation of the receptor stimulates Janus protein kinases, which in turn phosphorylate several proteins, including STAT1 and STAT2. The encoded protein also functions as an antiviral factor. [provided by RefSeq, Jul 2008]