We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
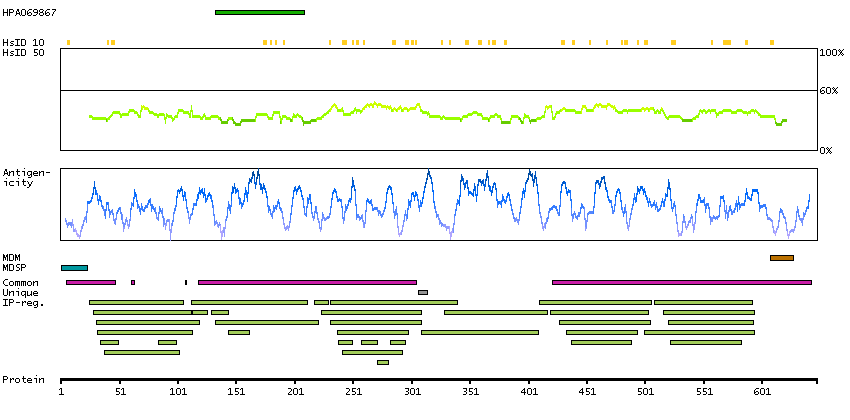
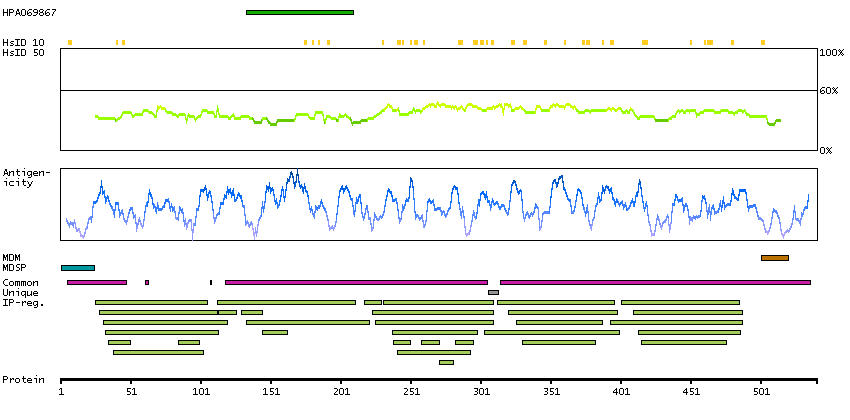
This gene is a member of the Ig superfamily and encodes a cell surface sialoglycoprotein expressed by cytokine-activated endothelium. This type I membrane protein mediates leukocyte-endothelial cell adhesion and signal transduction, and may play a role in the development of artherosclerosis and rheumatoid arthritis. Three alternatively spliced transcripts encoding different isoforms have been described for this gene. [provided by RefSeq, Dec 2010]