We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
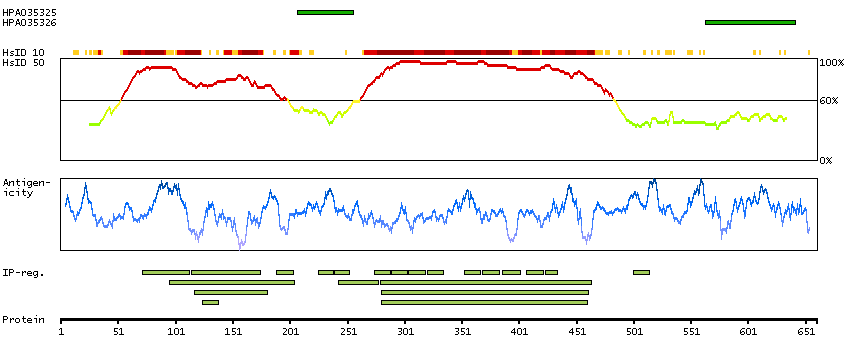
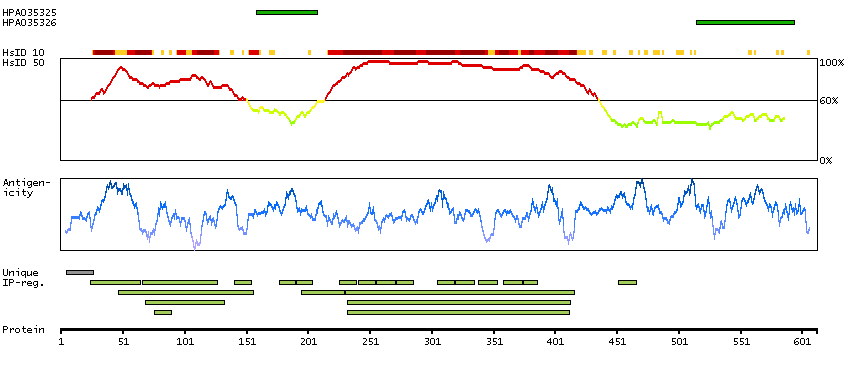
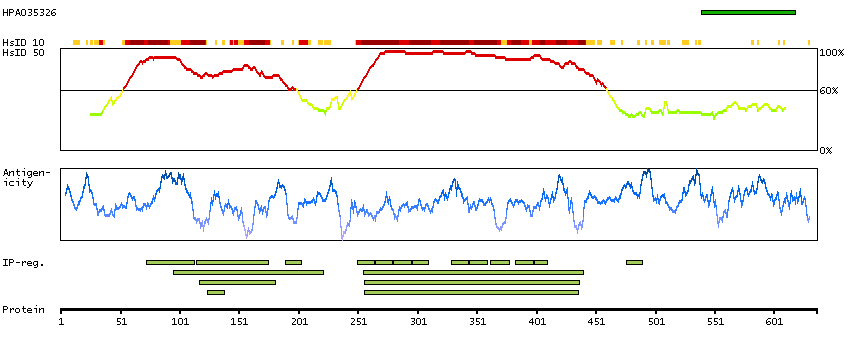
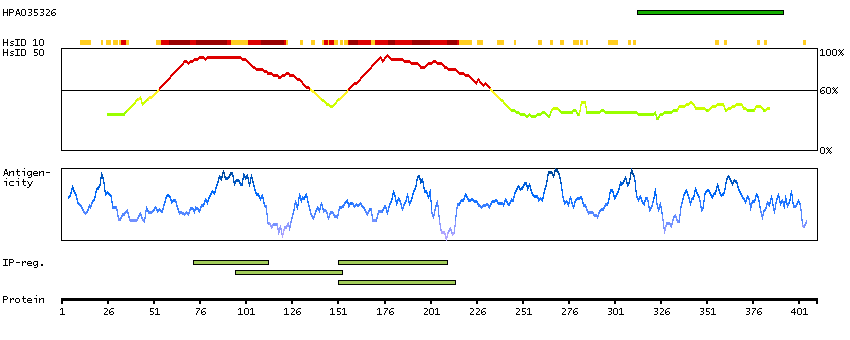
This gene encodes a subunit of a voltage-dependent calcium channel protein that is a member of the voltage-gated calcium channel superfamily. The gene product was originally identified as an antigen target in Lambert-Eaton myasthenic syndrome, an autoimmune disorder. Mutations in this gene are associated with Brugada syndrome. Alternatively spliced variants encoding different isoforms have been described. [provided by RefSeq, Feb 2013]
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)
Transporters Accessory Factors Involved in Transport Predicted intracellular proteins Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014)