We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
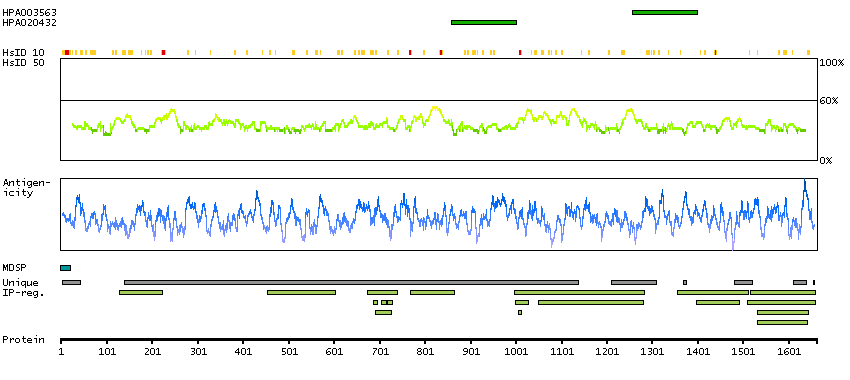
Complement component C3 plays a central role in the activation of complement system. Its activation is required for both classical and alternative complement activation pathways. The encoded preproprotein is proteolytically processed to generate alpha and beta subunits that form the mature protein, which is then further processed to generate numerous peptide products. The C3a peptide, also known as the C3a anaphylatoxin, modulates inflammation and possesses antimicrobial activity. Mutations in this gene are associated with atypical hemolytic uremic syndrome and age-related macular degeneration in human patients. [provided by RefSeq, Nov 2015]