We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (hippocampus, brain, caudate, cerebellum)
GENE INFORMATION
Gene name
GFAP (HGNC Symbol)
Synonyms
FLJ45472
Description
Glial fibrillary acidic protein (HGNC Symbol)
Entrez gene summary
This gene encodes one of the major intermediate filament proteins of mature astrocytes. It is used as a marker to distinguish astrocytes from other glial cells during development. Mutations in this gene cause Alexander disease, a rare disorder of astrocytes in the central nervous system. Alternative splicing results in multiple transcript variants encoding distinct isoforms. [provided by RefSeq, Oct 2008]
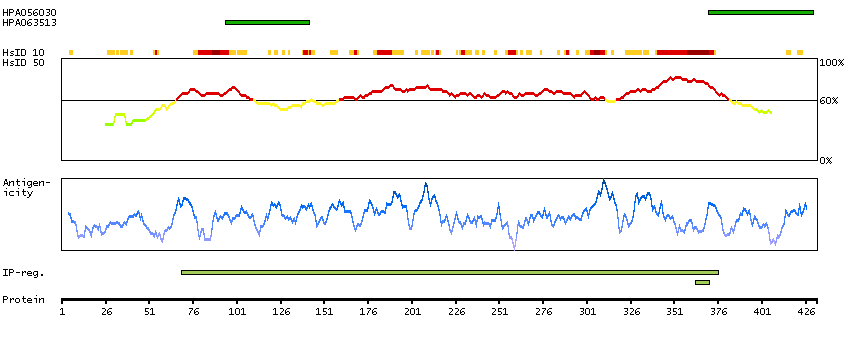
P14136 [Direct mapping] Glial fibrillary acidic protein
Show all
Predicted intracellular proteins Plasma proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
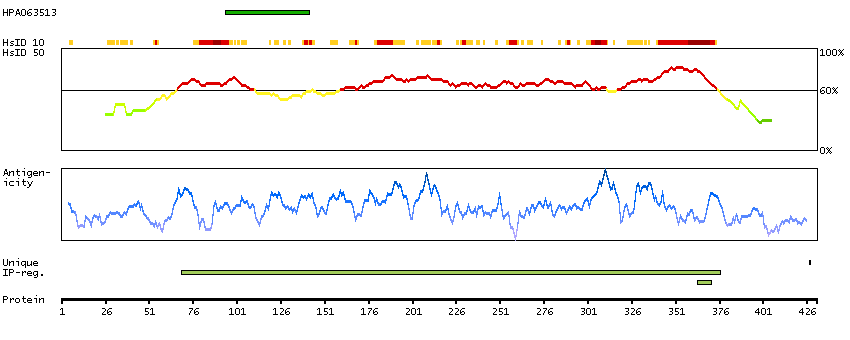
P14136 [Direct mapping] Glial fibrillary acidic protein
Show all
Predicted intracellular proteins Plasma proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
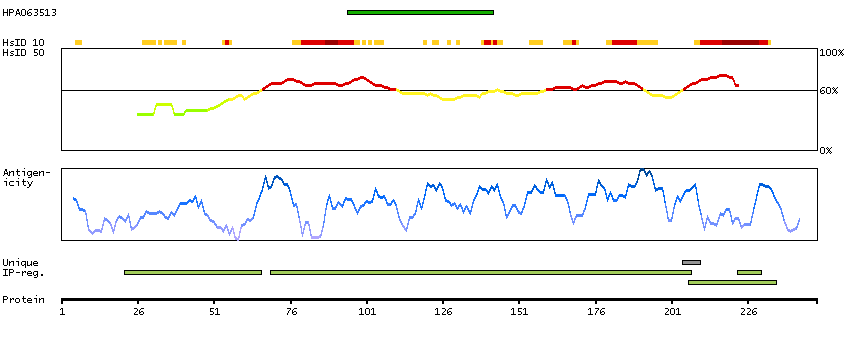
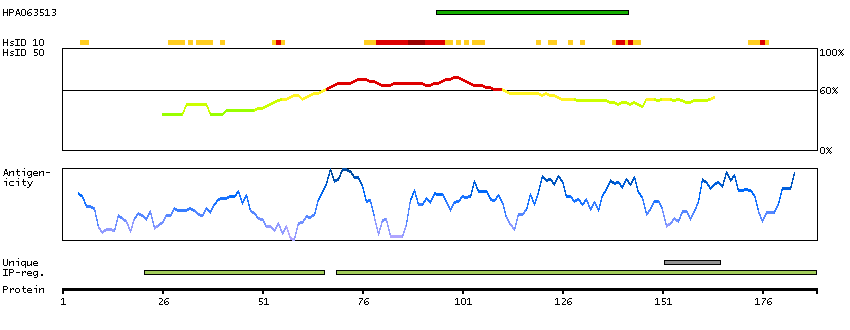
P14136 [Direct mapping] Glial fibrillary acidic protein
Show all
Predicted intracellular proteins Plasma proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)