We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
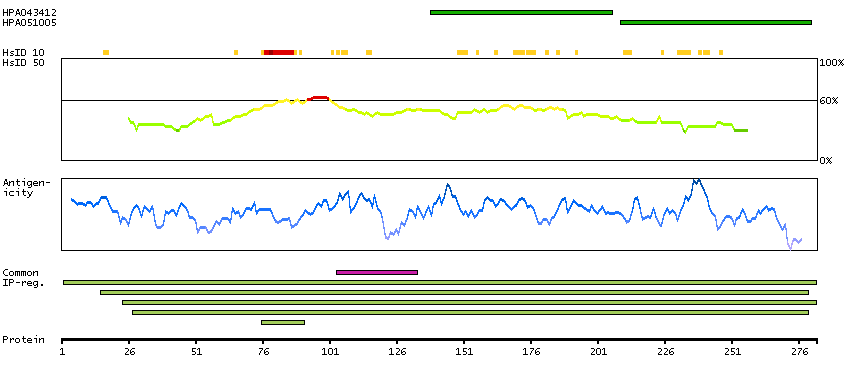
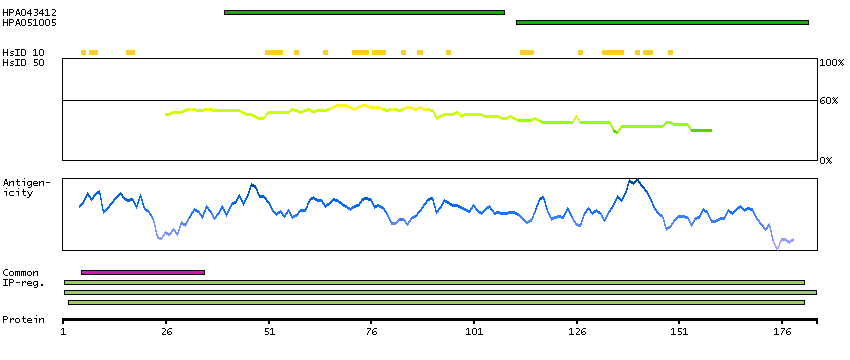
The product of this gene catalyzes the last step of the catecholamine biosynthesis pathway, which methylates norepinephrine to form epinephrine (adrenaline). The enzyme also has beta-carboline 2N-methyltransferase activity. This gene is thought to play a key step in regulating epinephrine production. Alternatively spliced transcript variants have been found for this gene. [provided by RefSeq, Nov 2012]
Enzymes ENZYME proteins Transferases Predicted intracellular proteins Plasma proteins Cancer-related genes Candidate cancer biomarkers Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)