We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cadherin 2, type 1, N-cadherin (neuronal) (HGNC Symbol)
Entrez gene summary
This gene encodes a classical cadherin and member of the cadherin superfamily. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein is proteolytically processed to generate a calcium-dependent cell adhesion molecule and glycoprotein. This protein plays a role in the establishment of left-right asymmetry, development of the nervous system and the formation of cartilage and bone. [provided by RefSeq, Nov 2015]
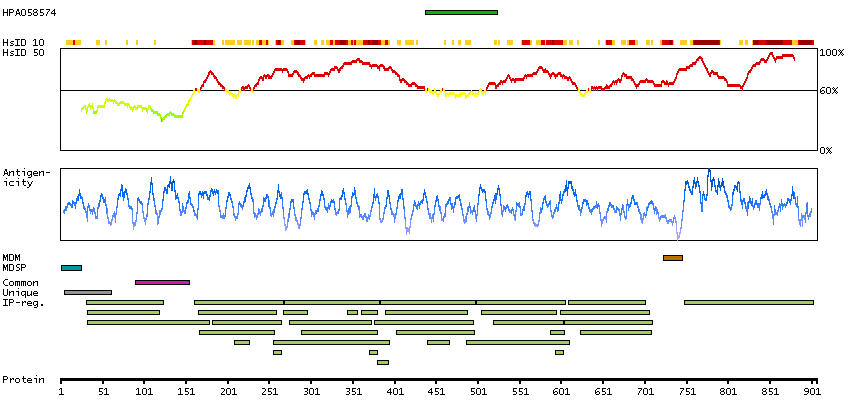
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005509 [calcium ion binding] GO:0007156 [homophilic cell adhesion via plasma membrane adhesion molecules] GO:0009966 [regulation of signal transduction] GO:0016020 [membrane]
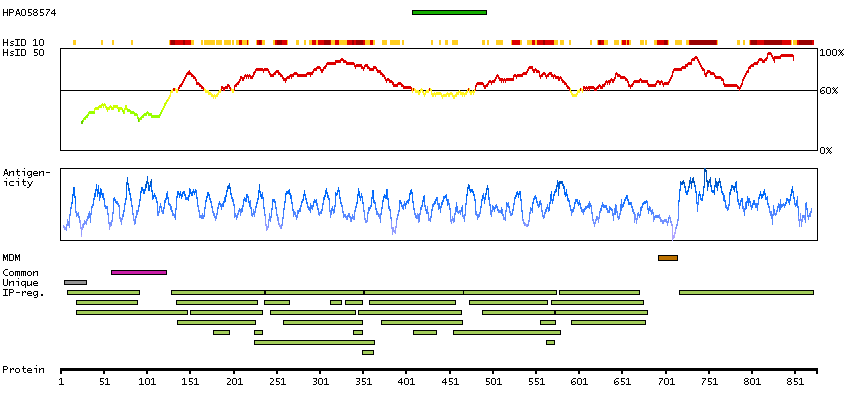
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005509 [calcium ion binding] GO:0007156 [homophilic cell adhesion via plasma membrane adhesion molecules] GO:0009966 [regulation of signal transduction] GO:0016020 [membrane]
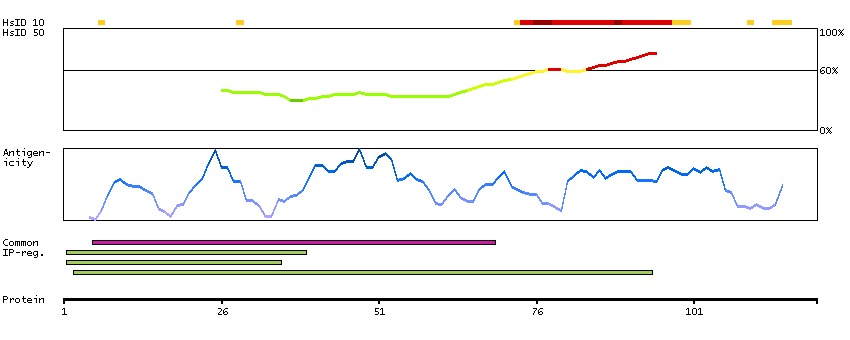
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005509 [calcium ion binding] GO:0007156 [homophilic cell adhesion via plasma membrane adhesion molecules] GO:0009966 [regulation of signal transduction] GO:0016020 [membrane]