We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
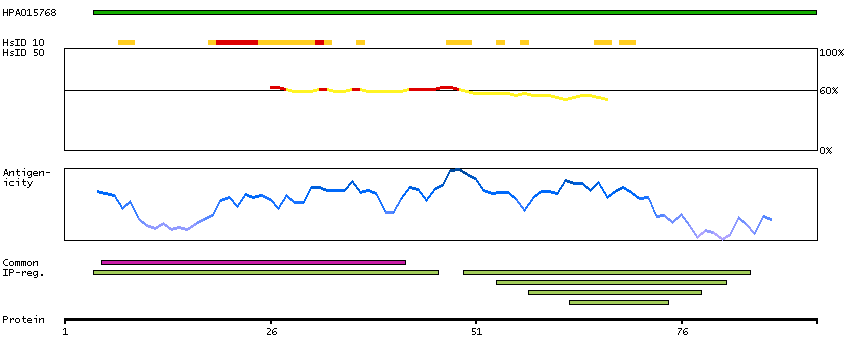
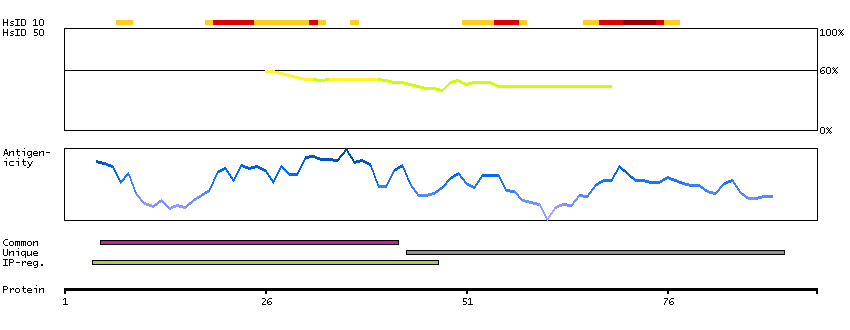
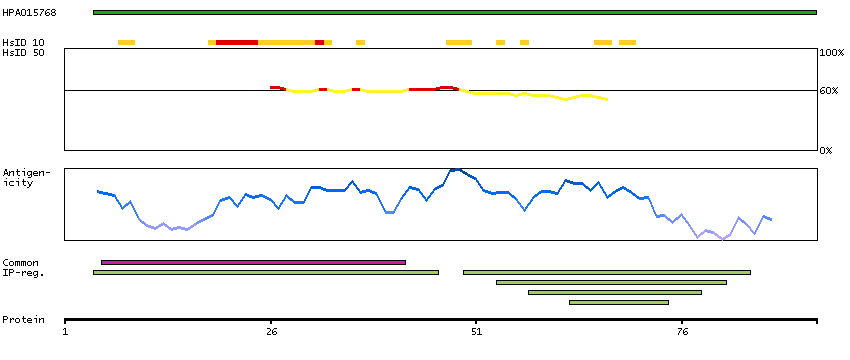
The protein encoded by this gene is a member of the S100 family of proteins containing 2 EF-hand calcium-binding motifs. S100 proteins are localized in the cytoplasm and/or nucleus of a wide range of cells, and involved in the regulation of a number of cellular processes such as cell cycle progression and differentiation. S100 genes include at least 13 members which are located as a cluster on chromosome 1q21; however, this gene is located at 21q22.3. This protein may function in Neurite extension, proliferation of melanoma cells, stimulation of Ca2+ fluxes, inhibition of PKC-mediated phosphorylation, astrocytosis and axonal proliferation, and inhibition of microtubule assembly. Chromosomal rearrangements and altered expression of this gene have been implicated in several neurological, neoplastic, and other types of diseases, including Alzheimer's disease, Down's syndrome, epilepsy, amyotrophic lateral sclerosis, melanoma, and type I diabetes. [provided by RefSeq, Jul 2008]
MEMSAT-SVM predicted membrane proteins SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers Candidate cardiovascular disease genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)